
蛋白质组学中质谱数据标准研究进展
2011-01-30 02:15马海滨张纪阳孙汉昌谢红卫
质谱学报 2011年3期
马海滨,张纪阳,2,刘 辉,2,孙汉昌,2,谢红卫
(1.国防科学技术大学,机电工程与自动化学院自动控制系,湖南长沙 410073;2.军事医学科学院,放射与辐射医学研究所,北京蛋白质组研究中心,蛋白质组学国家重点实验室,北京 102206)
在后基因组时代,蛋白质组学(proteomics)成为生命科学研究中的一个热点[1]。由于缺乏类似基因组 PCR(polymerase chain reaction)扩增的样品倍增方法,蛋白质组学研究对实验技术的要求更高,因此,重复实验、多实验平台互补、多策略互补等方法在蛋白质组学中更加重要。这些方法是提高蛋白质鉴定和定量结果覆盖率及重复性的重要手段。目前,蛋白质组学中使用的主要研究策略大都基于质谱实验与分析。质谱方法具有高通量和高灵敏度的特点,是蛋白质组学研究的一项支撑技术[2-3]。
由于质谱仪种类繁多,精度和性能差异较大,实验产出数据格式多样,实验数据难以整合[4],而后续质谱数据处理的目的却是要从实验产出的海量数据中完成数据的获取、处理、存储和解释。在目前的研究发展阶段,实验策略和数据分析方法种类繁多,而且还在不断提出新的数据分析方法,在不同分析策略中使用的算法也不尽相同,在不同分析流程中使用的数据格式繁多且大多互不兼容,部分质谱数据文件格式列于表1。这些因素给数据共享和交换带来困难,不利于分析结果的整合,与数据处理的目标背道而驰。因此,有必要对质谱数据处理中的数据格式问题进行研究。2004年以来,相继提出了多种开放式数据标准并得到不同程度的应用,初步缓解了目前面临的质谱数据格式兼容性的问题。
本工作对目前已有的质谱数据标准进行综述,介绍质谱数据标准的研究现状,比较各种数据标准的特点与优缺点,并展望质谱数据标准可能的发展方向。
1 质谱数据标准研究现状
目前,蛋白质组学中质谱数据标准的主要制定组织是HUPO-PSI(Human Proteome Organization-Proteomics Standards Initiative)[5],此外系统生物学研究所(Institute for System Biology,ISB)[6]和欧洲生物信息学研究所(European Bioinformatics Institute,EBI)[7]也参与了质谱数据标准的制定。
2002年,在华盛顿召开的 HUPO会议上成立了PSI,其主要目标就是要在蛋白质组学领域中为数据表示定义公共数据标准,以解决蛋白质组学研究中数据格式不统一的问题,实现数据的比较、交换和验证[8]。质谱数据公共标准和控制字(controlled vocabulary)的制定工作主要由PSI的MS(mass spectrometry)组织完成。由于可扩展标记语言[9](extensible markup language,XML)是一种与平台无关的结构性信息表示方法,因此当时以及后来制定的数据标准主要是基于XML格式的[10]。数据标准不仅要支持多种质谱实验技术,还必须能够存储与质谱实验有关的MIAPE[11-12](the minimum information about a proteomics experiment)信息。
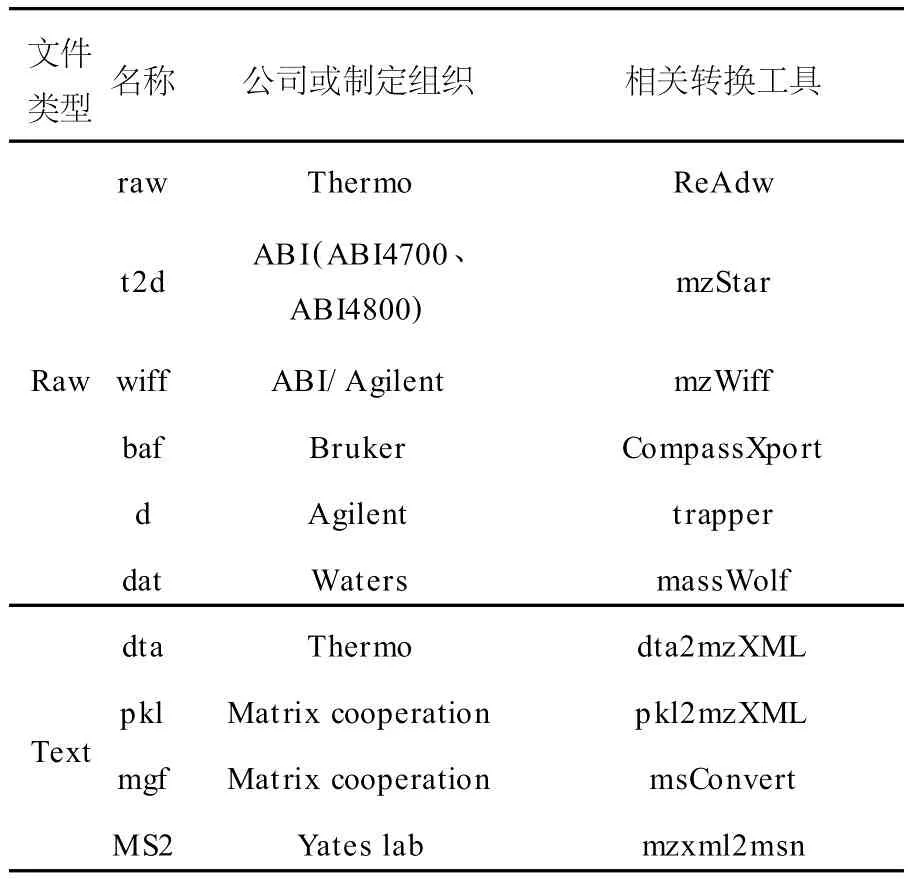
表1 质谱数据文件格式Table 1 File type of mass spectrometry data
2004年至2008年,用于存储和交换原始数据的标准主要有mzData与mzXML。2008年,PSI主持发布了 mzML,试图完全取代 mzData与mzXML。2003年,在蒙特利尔(Montreal)召开的 HUPO大会上,PSI-MS发布了 mzData 1.0版[13],目前使用的是2006年发布的mzData 1.05版。因其自身存在不足,该数据标准的应用相对比较有限。相对于mzData,ISB开发制定的数据标准 mzXML[14]得到了更广泛的应用。而在SPC(seattle proteome center)开发的数据分析平台 TPP(the trans-proteomic pipeline)中,使用的数据标准是 mzXML、pepXML和protXML,并试图兼容 mzML。2009年,PSI将AnalysisXML[15]中蛋白质鉴定部分的数据标准更名为mzIdentML,作为一个独立的数据标准发布,而关于蛋白质定量部分的数据标准暂时命名为mzQuantML[16]。另外,EBI在开发和维护数据库PRIDE(PRoteomics IDEntifications database)时,为其中的数据提供了一个专门的数据标准 Pride XML,该格式将 mzData整合其中,作为其保存质谱数据的数据格式,其余部分实现对搜库结果与实验信息的存储。表2列出了常用的数据标准及其制定组织,其中的数据分析阶段参照TPP中的划分。典型蛋白质鉴定流程示于图1。
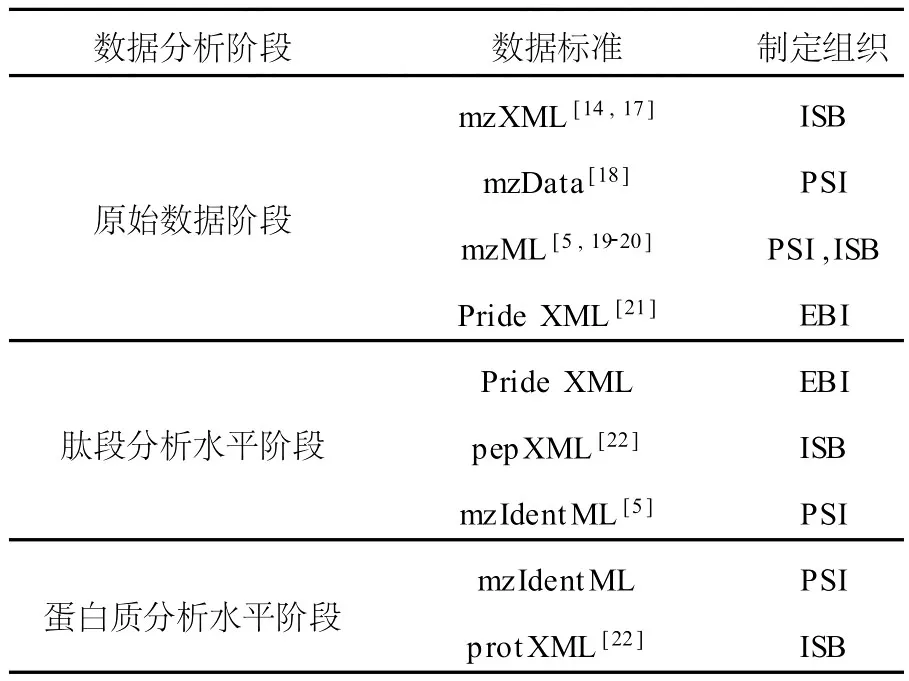
表2 现有质谱数据标准及其制定组织Table 2 Proteomics data format standards and corresponding organizations
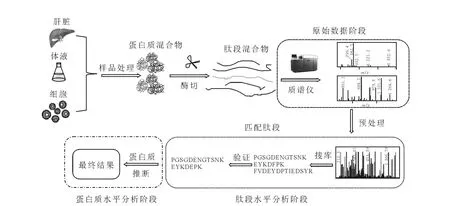
图1 典型蛋白质鉴定流程Fig.1 Typical workflow of protein identification
2 质谱数据标准介绍
蛋白质组学研究的一个重要工作就是蛋白质鉴定。在从质谱实验获得质谱数据,直到获得蛋白质鉴定结果的整个过程中,需要对不同格式的数据进行格式转换和数据整合,以满足不同数据分析软件的输入要求。在没有制定统一的数据标准之前,不统一的数据格式极大的限制了蛋白质组学数据的共享和发表,制约了相关数据库的开发,不利于研究人员对已有的研究成果进行再分析、整合和总结,导致不断的重复鉴定。随着研究工作的不断深入和研究成果的不断发表,构建蛋白质组学数据库就显得极为重要,而在整理数据时更需要一个统一的数据标准来对数据库中的数据进行管理[14,23-25]。
下面介绍基于质谱技术的蛋白质组学中常用的数据标准。在 TPP中,将数据处理过程划分为3个阶段:原始数据阶段、肽段水平分析阶段和蛋白质水平分析阶段[26]。本节将按照这3个阶段分别介绍各种数据标准。
2.1 原始数据阶段
该阶段数据标准的主要作用是将质谱仪生产商的原始数据格式转换为开放结构数据,以方便研究人员对数据的再次分析和对现有算法做出改进。这一阶段目前使用的数据标准主要有mzData,mzXML和mzML。其中,mzML是针对原始数据阶段最新制定的数据标准,它结合了前两种标准的优点。
2.1.1 mzData数据格式的特点 mzData的最大特点是使用XML模型外的控制字来描述与设备和实验设计等有关的参数。当采用新型仪器或新的实验方案时,这些参数能够以一个统一的数据格式存储在数据文件中。控制字可分成控制字参数(cvParam)和用户参数(userParam)两种。控制字参数具有一定的固定性,用户自定义的控制字可以放在用户参数中。使用控制字既保证了数据格式的可扩展性,又保证了数据格式的灵活性。但是,由于没有采取一定的机制限制用不同的方式编码本质相同的信息,导致同一版本之间控制字的不一致,严重影响了数据的共享以及读写软件的通用性。而且mzData中没有使用索引,不能实现对数据文件中质谱图的快速随机读取。
2.1.2 mzXML数据格式的特点 mzXML是用于存储和交换质谱数据的开放数据格式,具有很高的灵活性,且能存放多种类型的数据——从未经任何处理的数据到经过深度数据处理的数据(如质心化、峰检测等)。
在质谱实验分析中,使用统一的数据标准可以方便地将新型质谱仪加入到数据分析平台中,便于实验数据的交换和发表,能为新的数据分析工具的开发提供统一平台,因此mzXML从发布至今,已经得到广泛的应用。但相对于二进制文件而言,基于XML的数据文件还存在一些不足,主要有两点:一是将原始文件转换成基于XML的数据文件,文件容量会增加。现在高精度质谱仪在1 h内的数据产出量会超过1 GB,数据经转换后会带来一定的存储问题。而且,在XML文件中不能直接包含二进制数据,需要转换成人工可阅读的数据文件,这就不可避免地造成文件容量的增加;二是降低了数据文件中信息的读取速度[14]。虽然在mzXML文件中使用了索引,避免了数据文件必须从开始读到结尾的弊端,但还不能实现按条件对图谱和数据信息进行读取。分析mzXML文件结构还可以得出:该数据文件并不适合于计算,而且也没有存储与实验设计有关的参数信息[17]。
2.1.3 mzML的发展进程 2008年以前,主要有两大开放数据标准:mzData和 mzXML。两种标准处理的是相同的数据信息,这势必增加软件开发人员的负担。因此,mzData和mzXML的制定组织联合仪器生产厂商、数据分析人员和一些终端用户,在 HUPO-PSI的赞助下[27],开发了新的数据标准——mzML,最初定名为“dataXML”,其目的是要完全的取代前两种数据标准。
制定mzML的最初设计方案是,让mzML继承mzData和mzXML各自的优点,并借鉴这两种标准在实际使用时积累的经验。在实现之初,开发人员遵从下面的设计原则:1)保持数据标准的简洁;2)避免用不同的方法对同一信息进行编码;3)为了可编码一些新的重要信息,数据标准可以有一定的灵活性,但要保证标准的稳定性;4)继承 mzData和mzXML的特色,但在最初版本中不需要太多体现;5)利用现有资源尽快完成初始版本(mzML 1.0);6)编写读、写软件验证数据标准[28]。已发布的mzML主框架图示于图2。为了保证mzML能立即被广泛应用,2008年6月发布mzML1.0时即向用户提供了可以读、写和验证的软件。目前最新版本是2010年6月发布的mzML1.1.1。
2.1.4 3种数据标准的比较 mzData和mzXML最大的区别是数据文件灵活性的设计理念不同。mzData通过使用主体模型外部的控制字实现数据灵活性,这样可以保证实际的xsd模型在几年内保持不变。随着设备和软件的升级,只需不断地更新控制字即可实现数据标准的升级,也无需过多修改后续的分析软件。mzXML有一个严格的数据模型,大多数的数据信息都已被列举在模型中,当需要支持新的数据特征时,即使添加一个属性,均需要对数据模型进行修订,也要对分析软件进行相应的修改,造成连续发布的mzXML版本间多是相近的。但是,使用这种模型的优点是数据模型稳定性高,便于软件的实现和数据文档的验证[20]。
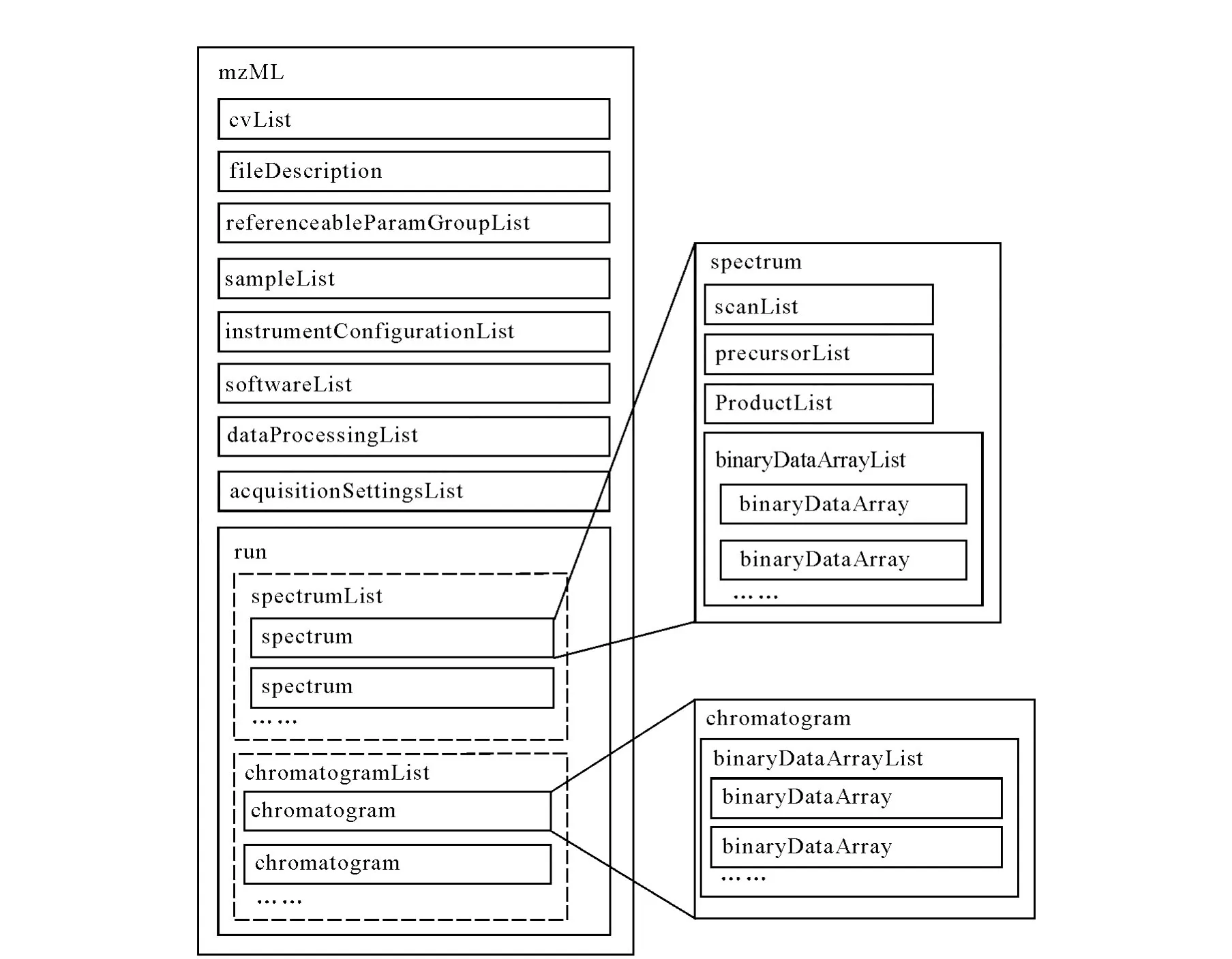
图2 mzML结构主框图Fig.2 The schema of mzML
mzML继承了上述两种格式各自的优点,使用控制字实现数据标准的灵活性,并使用控制字语法验证器,避免用不同的方法编码本质上相同的信息,以及各种版本间的不一致性,使mzML成为新一代基于 XML的开放式数据标准。
与mzXML相似,mzML使用索引来尝试解决质谱图随机读取的问题,但是该方法存在一定的风险。从mzXML多年的使用经验可以得出,索引技术带来的技术优势要远大于其带来的风险,但研究人员对在XML文件中引入索引方法褒贬不一。因此,一个*.mzML文件中可能会包含一个无索引的mzML文档或是带索引的mzML文档。
2.2 肽段水平分析阶段
在蛋白质鉴定流程中,一个典型的肽段匹配策略是数据库搜索[29],简称搜库。目前 TPP中支持的搜库工具有X!Tandem[30]、ProbID[31]、Mascot[32]、SEQUEST[33]、Phenyx[34]等 ,其中 ,X!Tandem和 ProbID是开源搜库软件。每个软件都有各自的数据输入输出文件格式,而且在实际分析中,需要用不同的软件对同一批数据进行搜库,不同格式的输出文件会对结果的比较、整合带来一定困难,不利于大规模的数据分析。因此,研究人员在这一阶段开发出的数据标准主要有:mzIdentML、pepXML和 Pride XML。其中,mzIdentML是针对该分析阶段最新制定的数据标准,适用于搜库后的数据分析流程。
2.2.1 mzIdentML的发展进程 2006年,PSIPI(proteomics informatics standards group)开始着手制定AnalysisXML的UML模型(unified modeling language model)[35]。随后在2008年的 PSI春季会议上,决定从 AnalysisXML中去除与定量有关的部分,主要是因为定量中使用的不同策略(有标定量和无标定量)以及新技术的不断更新,导致AnalysisXML1.0的发布一再推迟。2009年PSI的春季会议正式决定将AnalysisXML分成两部分:蛋白质鉴定数据标准——mzIdentML和蛋白质定量数据标准——mzQuantML[16]。mzIdentML继承了AnalysisXML的大部分内容,而mzQuantML还需要单独开发,但仍期望在开发时使其具有与mzIdentML高度相似的上层结构。2009年 8月,mzIdentML1.0版正式发布,而 mzQuant-ML仍在制定中。
mzIdentML主要包含 pepXML和protXML中的数据信息,以及其他一些相关信息(与定量无关的)。pepXML是 TPP在肽段水平数据分析中使用的数据标准;protXML是TPP在蛋白质水平数据分析中使用的数据标准。因此,在基于质谱的蛋白质鉴定中,mzI-dentML是搜库后结果的一个公共数据标准。TPP目前默认的数据标准还是pepXML,但在mzIdentML成熟后,TPP将会把最后的分析结果转化成mzIdentML格式数据[36]。
2.2.2 mzIdentML的结构特点 mzIdentML是对 FuGE(functional genomics experiment)对象模型的延伸,可以应用于 MS、MS/MS、MSn数据的搜库结果,比pepXML的适用性要广。
mzIdentML同样使用控制字来实现数据格式的灵活性,节点cvList中包含文件使用的控制字列表。节点Analysis Sample Collection使用控制字术语对试验中质谱仪分析的样本进行描述,若样本是混合样本,还需要定义其父样本。Sequence Collection分为DBSequence和Peptide两个子节点,前者为特定搜索数据库(核酸或氨基酸)中的一个数据库序列,后者为肽段序列(或修饰后序列),这两个序列作为搜索结果的参考序列集。对数据集进行图谱鉴定分析和蛋白质检测所需要的参数和设置均存放在子节点Analysis Protocol Collection中,而分析过程中得到的数据结果存储在 AnalysisData中。mzIdentML支持对数据的多次搜索,搜索结果可以存储在同一个数据文件中。
mzIdentML数据标准适用于肽段水平分析和蛋白质水平分析,为整个蛋白质鉴定过程减轻了工作量,有效地避免了在分析中对不同数据格式进行转换。
2.2.3 pepXML的结构特点 pepXML是ISB开发的用来存储、交换和处理肽段序列匹配数据的数据标准,仅适用于MS/MS的搜库结果。
在pepXML模型中,有相应的节点保证模型能支持搜库、结果验证和定量分析。其中,msms_pipeline_analysis存储与搜库有关的信息及搜库结果;peptideprophet_summary和peptideprophet_result存储与搜库结果验证有关的信息;asapratio_summary和asapratio_result存储与ASAPRatio有关的定量信息。
pepXML支持在单文档中存储多次搜库结果。每一次搜库的结果都放在msms_run_summary中,其中包含原mzXML文件的信息和从mzXML文件得到质谱仪的详细描述以及在试验中使用的水解酶信息。此外还包含一个search_summary子节点,其中存储与搜索引擎和搜索数据库、肽段修饰、酶和序列搜索限制配置有关的信息,并将图谱名、母离子电荷和质量以及节点search_result放在其子节点spectrum_query中。在对同一数据集做多次搜索时,每个search_result通过唯一的search_id与其对应的search_summary相连接,以实现在单文件中存储多次搜索结果。
pepXML中还包含搜库结果验证和定量的模块,如 TPP中使用的结果验证软件 PeptideProphet和定量软件 XPRESS[37]、ASAPRatio[38],在pepXML中均有对应的数据模块。
与SEQUEST的输出文件SQL相比,pep-XML是一个有严格模型的XML文件格式,便于数据使用者验证数据文件格式是否正确,保证分析软件能有一个可靠的输入数据。
2.2.4 Pride XML的结构特点 Pride XML是 EBI为数据库PRIDE[39-41]专门开发的一个数据格式,其中包含了完整的图谱数据以及搜库结果。该标准在图谱数据方面完全使用了mzData数据标准,将其作为一个节点 mzData。Pride XML中可以不包含mzData格式的图谱数据,但在mzData节点中必须要对实验样品、仪器设备和数据处理软件等参数进行详细的描述。
Pride XML将基于不同实验的搜库结果分别存放在不同的节点中,二维凝胶电泳的搜库结果存储在 TwoDimensionalIdentification中,其它方法的搜库结果存储在 GelFreeIdentification中。同一肽段序列(或是重叠序列)的搜索结果存放在节点 GelFreeIdentification中,其中还需要包含的信息有搜索数据库的名称及版本、搜索引擎名称及版本、相关图谱ID、各项搜库结果值以及修饰属性等。
Pride XML同样利用控制字来实现数据格式的灵活性,用户可以在PRIDE的网站上对自己的XML文件进行验证。
2.3 蛋白质水平分析阶段
蛋白质鉴定过程的最后一步是通过搜库中获得的肽段结果推断出样品中含有的蛋白质。这一阶段中已有的数据标准有mzIdentML和protXML,前者已在上一节做了介绍。
从得到的肽段序列推断出蛋白质序列的方法有多种,不同的方法有不同格式的结果文件。protXML即是一个用于存储、交换和处理基于串联质谱的蛋白质鉴定结果的开放式数据标准。
protXML的节点protein_summary_header包含与肽段鉴定相关的信息,一个包含蛋白质鉴定方法信息的子节点program_details和一个包含该方法细节信息的通配符。蛋白质鉴定结果存放在protein_group中,可以有多组,每组中均有一个编号group_number,每组中还可以包含一个或多个protein节点。protXML也适用于鉴定结果的后续蛋白质水平上的分析,如XPRESS和 ASAPRatio蛋白质定量。protXML文件还分别设有对应的节点存储相应的分析结果,如ASAPRatio的定量结果存储在节点ASAPRatio中。
3 支持数据标准的软件
上述数据标准自发布之日起便逐步得到了应用,常用的蛋白质组质谱分析软件通常都支持多种数据标准,表3给出了常用软件对数据标准的支持情况。
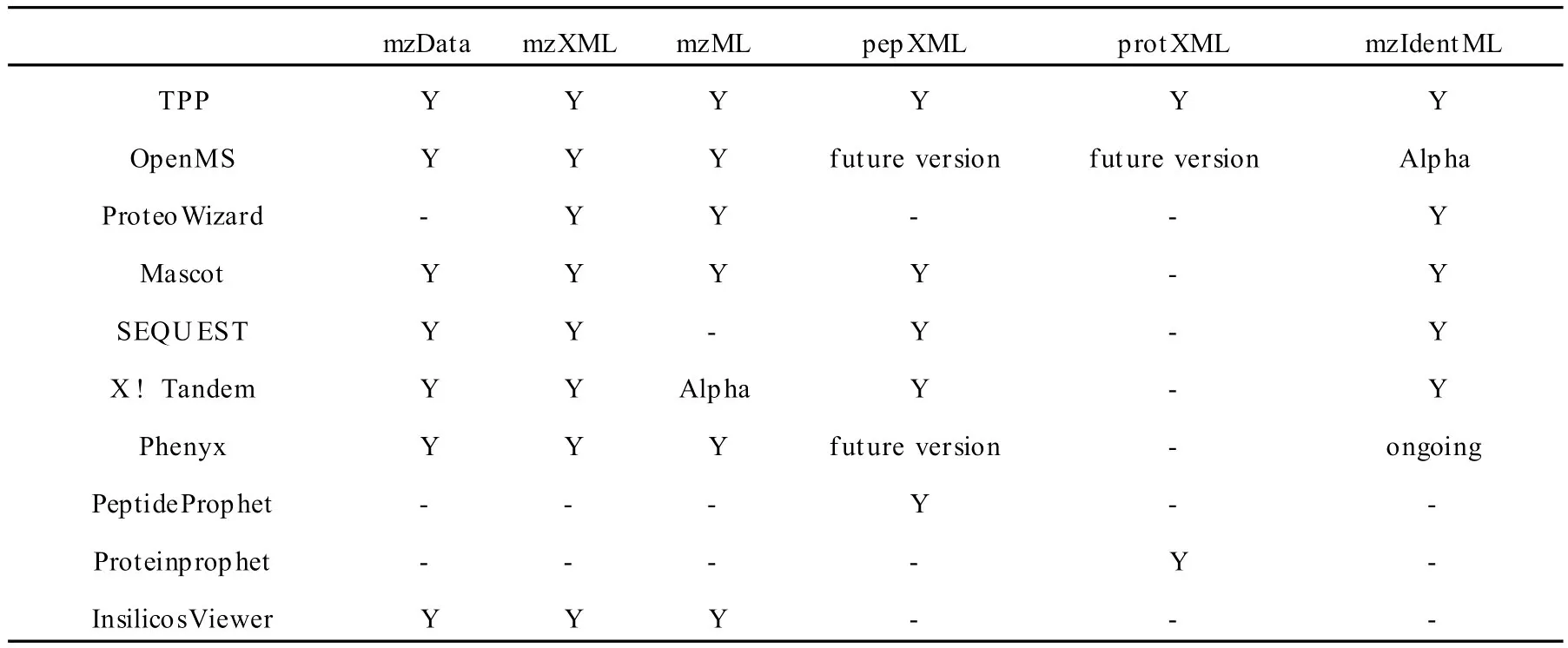
表3 常用支持数据标准的软件列表Table 3 Widely-used softwares and their support for the XML-based data format standards
4 基于XML数据标准的优缺点
基于XML的质谱数据标准在质谱数据分析中发挥了极大的作用,减轻了分析人员在数据分析中对不同格式的数据进行转换的工作,有利于大规模数据分析平台的建设。但这些数据标准还存在一定的问题,需要不断的完善和发展。
首先,已有标准对原始数据共享的支持力度比较大,但是对数据分析结果的支持有限。ISB制定的pepXML和protXML,以及 EBI的Pride XML虽然提供了部分对搜库结果存储的支持,但是还不能兼容所有出现的、典型的数据分析流程。
其次,这些数据标准多以灵活的XML格式实现,其中大量的数据标签使容量本已足够庞大的高通量质谱数据文件更加庞大,数据压缩虽然可以部分解决这个问题[42-43],但是不能兼顾数据的访问速度。
最后,XML平坦式的存储方式给数据的高效访问带来了一定负担,通过建立额外的索引虽可以部分解决这一问题,但是直接的索引式存储效率应更加高效。
5 展望
质谱数据具有数据量大和数据格式不统一的特点,研究工作的需求,牵引和推动了数据标准的发展,但目前数据标准的制定仍然落后于数据的发展。通过比较目前已有的数据标准并结合实际数据处理的诸多经验,建议从两个不同的层面关注和推动今后数据标准的发展:
1)对数据标准的制定组织,仍需继续完善和发展蛋白质组学中所需数据标准。上文介绍的很多数据标准仅仅适用于蛋白质鉴定的典型流程,使用范围有限。因此,数据标准的制定还需要不断完善。从各数据标准的发展过程以及mzML与mzIdentML的制定过程可以推断出质谱数据标准在向一个可以支持所有蛋白质组学典型分析流程的方向发展。由于蛋白质组学自身的特点,如实验策略多、分析步骤复杂以及数据分析算法繁多等,使得这一发展过程困难重重。
2)对数据分析人员,在目前数据标准不统一的状况下,可开发基于已有数据标准的适用于典型蛋白质鉴定流程的数据格式,并充分利用已有的开源数据格式转换工具,将当前各种不同数据标准的数据信息合理地整合到一个统一的数据格式中,以简化数据分析流程。在此实践过程中,可以向数据标准制定组织及时反馈使用经验,共同推动蛋白质组学中数据标准的发展。
[1]Science Editor.Breakthrough of the year.Peering into 2002[J].Science,2001,294(5 551):2 444.
[2]AEBERSOLD R,MANN M.Mass spectrometrybased proteomics[J].Nature,2003,422(6 928):198-207.
[3]ONG S E,MANN M.Mass spectrometry-based proteomics turns quantitative[J].Nat Chem Biol,2005,1(5):252-262.
[4]STATES D J,OMENN G S,BLACKWELL T W,et al.Challenges in deriving high-confidence protein identifications from data gathered by a HUPO plasma proteome collaborative study[J].Nat Biotechnol,2006,24(3):333-338.
[5]The HUPO Proteomics Standards Initiative[OL][2010].http://www.psidev.info/.
[6]Institute for System Biology[OL][2010].http://www.systemsbiology.org/.
[7]European Bioinformatics Institute[OL][2010].http://www.ebi.ac.uk/.
[8]KA ISER J.Proteomics:Public-private group maps outinitiatives[J]. Science,2002,296(5 569):827.
[9]ACHARD F,VAYSSEIX G,BARILLOT E.XML,bioinformatics and data integration[J].Bioinformatics,2001,17(2):115-125.
[10]BRAY T,PAOLI J,SPERBERG-MCQUEEN C M,et al.Extensible Markup Language(XML)1.0(Second Edition),2000.
[11]TAYLOR C F,PATON N W,LILLEY KS,et al.The minimum information about a proteomics experiment(MIAPE)[J].Nat Biotechnol,2007,25(8):887-893.
[12]TAYLOR C F,BINZ P A,AEBERSOLD R,et al.Guidelines for reporting the use of mass spectrometry in proteomics[J]. NatBiotechnol,2008,26(8):860-861.
[13]ORCHARD S,TAYLOR C F,HERMJAKOB H,et al.Advances in the development of common interchange standards for proteomic data[J].Proteomics,2004,4(8):2 363-2 365.
[14]PEDRIOLI P G,ENGJ K,HUBLEY R,et al.A common open representation of mass spectrometry data and its application to proteomics research[J].Nat Biotechnol,2004,22(11):1 459-1 466.
[15]VIZCAINO J A,MARTENS L,HERMJAKOB H,et al.The PSI formal document process and its implementation on the PSI website[J].Proteomics,2007,7(14):2 355-2 357.
[16]ORCHARD S,DEUTSCH E W,BINZ P A,et al.Annual spring meeting of the Proteomics Standards Initiative[J].Proteomics,2009,9(19):4 429-4 432.
[17]LIN S M,ZHU L,WINTER A Q,et al.What is mzXML good for?[J].ExpertRev Proteomics,2005,2(6):839-845.
[18]PSI-MS:Mass Spectrometry Standards Working Group[OL][2010].http://www.psidev.info/index.php?q=node/80.
[19]ORCHARD S,MONTECHI-PALAZZIL,DEUTSCH E W,et al.Five years of progress in the standardization of proteomics data 4th annual spring workshop of the HUPO-proteomics standards initiative April 23-25,2007 ecolenationalesuperieure(ENS),Lyon,France[J].Proteomics,2007,7(19):3 436-3 440.
[20]DEU TSCH E.mzML:A single,unifying data format for mass spectrometer output[J].Proteomics,2008,8(14):2 776-2 777.
[21]PRoteomics IDEntifications database(PRIDE)[J/OL][2010].http://www.ebi.ac.uk/pride/.
[22]Seattle proteome center(SPC)-Proteomics Tools[EB/OL].http://tools.proteomecenter.org/software.php.
[23]PRINCE J T,CARL SON M W,WAN G R,et al.The need for a public proteomics repository[J].Nat Biotechnol,2004,22(4):471-472.
[24]CARR S,A EBERSOLD R,BALDWIN M,et al.The need for guidelines in publication of peptide and protein identification data:Working group on publication guidelines for peptide and protein identification data[J].Mol Cell Proteomics,2004,3(6):531-533.
[25]ORCHARD S,HERMJAKOB H,JULIAN R K,et al.Common interchange standards for proteomics data:Public availability oftools and schema[J].Proteomics,2004,4(2):490-491.
[26]KELL ER A,ENGJ,ZHANG N,et al.A uniform proteomics MS/MS analysis platform utilizing open XML file formats[J].Mol Syst Biol,2005:0017.
[27]ORCHARD S,HERMJAKOB H.The HUPO proteomics standards initiative-easing communication and minimizing data loss in a changing world[J].Brief Bioinform,2008,9(2):166-173.
[28]DEUTSCH E W.Mass spectrometer output file format mzML[J].Methods Mol Biol,2010,604:319-331.
[29]XU C,MA B.Software for computational peptide identification from MS-MS data[J].Drug Discov Today,2006,11(13/14):595-600.
[30]CRAIG R,BEAVIS R C.TANDEM:Matching proteins with tandem mass spectra[J].Bioinformatics,2004,20(9):1 466-1 467.
[31]ZHANG N,AEBERSOLD R,SCHWIKOWSKI B.ProbID:A probabilistic algorithm to identify peptides through sequence database searching using tandem mass spectral data[J].Proteomics,2002,2(10):1 406-1 412.
[32]PERKINS D N,PAPPIN D J,CREASY D M,et al.Probability-based protein identification by searching sequence databases using mass spectrometry data[J]. Electrophoresis,1999,20(18):3 551-3 567.
[33]ENGJ K,MCCORMACK A L,IIIJRY.An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database[J].Journal of the American Society for Mass Spectrometry,1994,5(11):976-989.
[34]COLINGE J,MASSELOT A,GIRON M,et al.OLAV:Towards high-throughput tandem mass spectrometry data identification[J].Proteomics,2003,3(8):1 454-1 463.
[35]ORCHARD S,APWEILER R,BARKOVICH R,et al.Proteomics and Beyond:A report on the 3rd annual spring workshop of the HUPO-PSI 21-23 April 2006,San Francisco,CA,USA[J].Proteomics,2006,6(16):4 439-4 443.
[36]DEUTSCH E W,MENDOZA L,SHTEYNBERG D,et al.A guided tour of the trans-proteomic pipeline[J].Proteomics,2010,10(6):1 150-1 159.
[37]HAN D K,ENGJ,ZHOU H,et al.Quantitative profiling of differentiation-induced microsomal proteins using isotope-coded affinity tags and mass spectrometry[J].Nat Biotechnol,2001,19(10):946-951.
[38]LI XJ,ZHANG H,RANISH J A,et al.Automated statistical analysis of protein abundance ratios from data generated by stable-isotope dilution and tandem mass spectrometry[J].Anal Chem,2003,75(23):6 648-6 657.
[39]MARTENS L,HERMJA KOB H,JONES P,et al.PRIDE:The proteomics identifications database[J].Proteomics,2005,5(13):3 537-3 545.
[40]JONES P,COTE R G,MARTENS L,et al.PRIDE:A public repository of protein and peptide identifications for the proteomics community[J].Nucleic Acids Res,2006,34(suppl 1):D659-D663.
[41]JONES P,COTE RG,CHO S Y,et al.PRIDE:New developments and new datasets[J].Nucleic Acids Res,2008,36(suppl 1):D878-D883.
[42]MIGUEL A C,KEANE J F,WHITEAKER J,et al.Compression of LC/MS Proteomic data[C].19th IEEE Symposium on Computer-Based Medical Systems, Salt Lake City, 2006:925-930.
[43]MIGUEL A C,KEARNEY-FISCHER M,KEANE J F,et al.Near-lossless compression of mass spectra for proteomics[C].Acoustics,Speech,and Signal Processing,Honolulu,2007:1 369-1 372.
猜你喜欢
昆明医科大学学报(2022年3期)2022-04-19
食品安全导刊(2021年20期)2021-08-30
昆明医科大学学报(2021年4期)2021-07-23
天然产物研究与开发(2018年2期)2018-04-04
科学与财富(2017年33期)2017-12-19
电脑知识与技术(2017年6期)2017-04-26
科技与创新(2016年17期)2016-11-04
医学研究杂志(2015年11期)2015-06-10
特产研究(2014年4期)2014-04-10
中国环境科学(2014年4期)2014-02-02
