
甲醇在FeS2(100)完整表面的吸附和分解
2011-11-30 10:41杜玉栋赵伟娜章永凡陈文凯
物理化学学报 2011年5期
杜玉栋 赵伟娜 郭 欣 章永凡 陈文凯,*
(1福州大学化学系,福州350108; 2华中科技大学煤燃烧国家重点实验室,武汉410074)
甲醇在FeS2(100)完整表面的吸附和分解
杜玉栋1赵伟娜1郭 欣2章永凡1陈文凯1,*
(1福州大学化学系,福州350108;2华中科技大学煤燃烧国家重点实验室,武汉410074)
采用基于第一性原理的密度泛函理论结合周期平板模型方法,研究了甲醇分子在FeS2(100)完整表面的吸附与解离.通过比较不同吸附位置的吸附能和构型参数发现:表面Fe位为有利吸附位,甲醇分子通过氧原子吸附在表面Fe位,吸附后甲醇分子中的C―O键和O―H键都有伸长,振动频率发生红移;甲醇分子易于解离成甲氧基CH3O和H,表面Fe位仍然是二者有利吸附位.通过计算得出甲醇在FeS2(100)表面解离吸附的可能机理:甲醇分子首先发生O―H键的断裂,生成甲氧基中间体,继而甲氧基C―H键断裂,得到最后产物HCHO和H2.
密度泛函理论;甲醇;FeS2(100)表面;吸附;过渡态
1 引言
矿物质表面在腐蚀、风化,重金属吸附及催化过程中具有重要的作用.二硫化亚铁作为地球表面最丰富的硫矿物之一,其表面活性涉及到废水处理,煤矿去硫等众多领域.作为吸附剂,它对海底沉淀物中微量元素的循环起到一定作用.1煤炭燃烧过程中,由于FeS2的存在会产生大量的SO2等有毒气体.人们研究用泡沫浮选技术除去煤炭中的FeS2,王淀佐等人2做了硫化矿的氧化与浮选机理的量子化学研究.有机小分子作为表面活性剂能够促进FeS2与煤炭的分离,3-5表面活性剂与FeS2表面的相互作用是表面活性剂的官能团与FeS2表面的化学吸附作用.此外,硫铁矿的氧化分解所产生的硫化物及有毒重金属会给地球带来巨大的环境污染,如酸性矿物质水.6-8人们通过让其吸附惰性官能团,如有机酸磷酸盐等来减少其氧化分解的速率.3,9其次FeS2的禁带宽度合适(0.95 eV),光吸收系数高(当波长λ<1 μm时,吸收系数α>105cm-1),可制作极薄的太阳能薄膜电池,被认为是一种极具发展潜力的太阳能电池材料.10
人们利用X射线光电子能谱(XPS),11-17扫描隧道显微镜(STM)18以及密度泛函理论(DFT)19-26研究了FeS2的电子结构,表面性质以及小分子在其表面的吸附机理.结果发现,FeS2(100)表面为其最稳定的表面,基本不发生弛豫与重构.Guevremont等18利用光电子吸附氙(PAX),程序升温脱附(TPD)及电子能量损失能谱(EELS)研究了H2O分子及甲醇分子在FeS2(100)表面的吸附与解离,结果发现,甲醇解离后有O原子与表面Fe成键.
本文通过密度泛函理论结合周期平板模型,研究了甲醇分子在FeS2(100)表面的吸附方式、相互作用机理及解离可能过程,从而为理解FeS2(100)表面与界面奠定基础.
2 理论模型和计算方法
2.1 理论模型
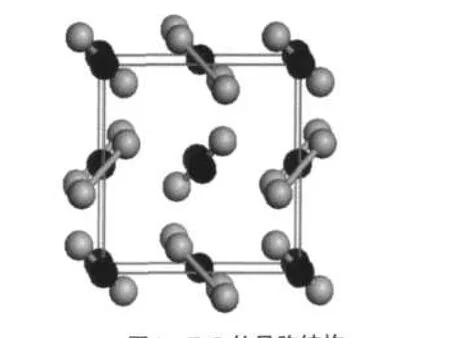
图1 FeS2的晶胞结构Fig.1 Unit cell of the pyrite structureGrey and black spheres represent S and Fe atoms,respectively.
FeS2结构类似于NaCl晶体结构,晶胞参数a= 0.5416 nm,27Fe原子位于单胞的角顶和面心,哑铃型的S原子对位于立方体单胞的12条棱上.由于单胞中哑铃状的S原子具有不同的或相反的取向,使其对称性从NaCl的面心立方Fm3m空间群变为Pa3空间群.具体构型如图1所示.
本文采用如图2所示的(2×2)超晶胞9层平板模型来模拟FeS2(100)表面.体相Fe原子和S原子分别为六配位和四配位,而表面Fe原子和S原子分别为五配位和三配位.计算时超晶胞表面三层原子放开自由度,下面六层固定.应用此模型成功计算了CO在其表面的吸附.28
2.2 计算方法
采用广义梯度密度泛函理论29和周期平板模型方法30模拟甲醇在FeS2(100)面的吸附和解离.所有的计算均由Dmol3软件包31,32实现.计算采用PBE泛函,Fe原子和S原子内层电子由有效核电势(ECP)代替,C、H和O采用全电子基组.价电子波函数采用双数值基加极化函数(DNP)展开,计算过程精度设为精细,优化收敛精度取程序内定值.相邻两层平板间的真空层厚度为1.2 nm.以确保平板间相互作用足够小.过渡态搜索采用Dmol3程序包中的com-plete LST/QST方法.采用相同方法优化气相的甲醇,构型参数分别为:dC-O=0.1447 nm,dC-H=0.1100 nm,dO-H=0.0989 nm,θHCH=109.5°,θCOH=109.1°分别与实验值0.1425 nm、0.1094 nm、0.0945 nm、108.6°和108.5°33相符合.
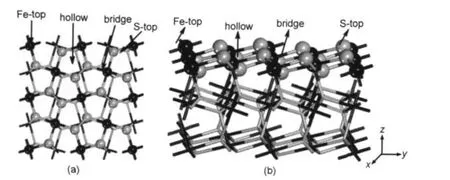
图2 超晶胞模型的俯视图(a)和侧视图(b)Fig.2 Top view(a)and side view(b)for the slab model
吸附能定义为吸附前后各物质总能量的变化: Eads=Emethanol+Esubstrate-Eadsorptionsystem,其中Emethnaol和Esubstrate分别表示吸附前甲醇分子和底物的能量,Eadsorptionsystem表示吸附后体系的总能量.
3 结果和讨论
3.1 甲醇在FeS2(100)表面的吸附
3.1.1 吸附构型和吸附能
根据FeS2(100)表面和甲醇分子的结构,甲醇分子分别以C―O键垂直和平行两种取向吸附.在FeS2(100)面选择Fe-top、S-top、bridge和hollow共四种吸附位,如图2所示.其中Fe-top和S-top吸附模式是指吸附分子分别以两种取向位于表面Fe和S原子的上方,吸附分子位于Fe―S键上方的吸附模式称为bridge,位于Fe―S形成穴位上方的称为hollow模式.本文用Vert-和Para-分别表示C―O键垂直和平行吸附于表面.
构型优化后,发现所有不同吸附位的构型共转化为两种构型,如图3所示.其中稳定构型为Vert-Fe-top(图3(a)),另一种为不稳定构型Para-top (图3(b)),吸附分子悬于底物上方.在稳定构型中,甲醇分子以C―O键倾斜吸附于表面,通过O原子与底物表面Fe成键,与实验结果18一致.Eads=69.3 kJ· mol-1,略高于实验值(46.2 kJ·mol-1),34属于弱的化学吸附.具体的构型参数列于表1,由于Vert-Fe-top为最稳定构型,表中只列出其构型参数.由表可见,甲醇吸附后整个分子结构的变化不大,C―O键由0.1425 nm伸长到0.1447 nm,O―H键伸长了0.0044 nm,C―H键只伸长了0.0006 nm,角度θCOH增大了1.6°左右.由此可见,甲醇与FeS2(100)表面之间的相互作用很弱.
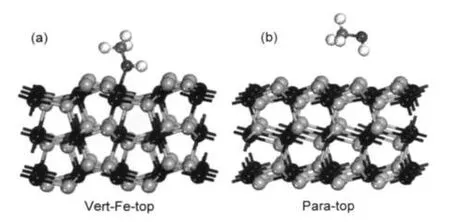
图3 甲醇分子在FeS2(100)面的两种吸附构型Fig.3 Two adsorption structures of methanol on FeS2(100)surface

表1 甲醇在FeS2(100)面吸附的吸附能和几何构型参数Table 1 Adsorption energy and geometrical parameters for methanol adsorbed on FeS2(100)surface
3.1.2 Mulliken电荷布居和轨道分析
甲醇在FeS2(100)表面吸附后的Mulliken电荷布居列于表2,吸附后甲醇带正电荷.C原子吸附后2s和2p轨道得电子,O原子2s和2p轨道失电子,H原子失电子;吸附位的Fe2+的4s和4p轨道得电子, 3d轨道失去0.04个电子,净结果为甲醇通过O原子把电子转移给底物.由此可见:H原子转移电子给C原子,C原子又把电子部分转移给O原子,O原子把得到的电子转移给Fe2+.结果与甲醇分子在Cu(111)35及Pt-Fe(111)/C36表面吸附过程相似.
在甲醇分子中,C和O都采取sp3杂化,O原子带有两对孤电子.甲醇分子的HOMO是n轨道,处于全充满状态,其主要成分来自于O原子的孤对电子;LUMO是σ*轨道,能量较高,无法从表面接受电子.37甲醇主要通过O原子的孤对电子与底物作用.表面五配位的Fe原子最高占据轨道主要是eg轨道,最低空轨道也包含eg轨道.38由于甲醇分子中O的p轨道与底物的Fe原子的eg轨道不相匹配,相互之间成键能力较弱,所以甲醇与底物之间的作用不强,吸附能不高,为弱的化学吸附.
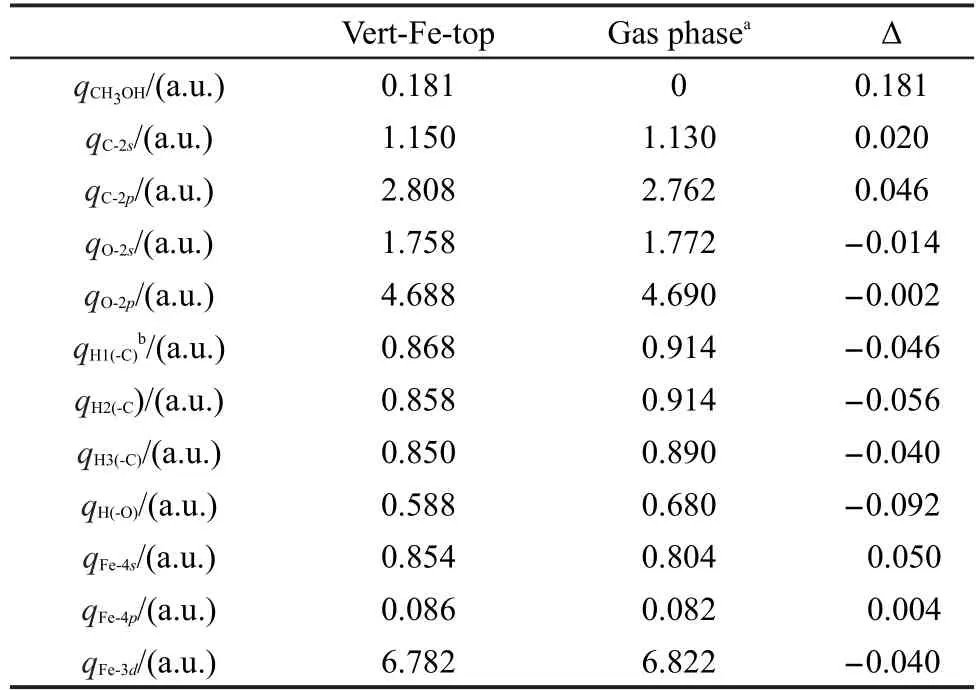
表2 甲醇各原子和Fe2+吸附前后的轨道电荷布居Table 2 Orbital charge populations for atoms of methanol and Fe2+before and after adsorption

表3 甲醇吸附前后振动频率的实验值与计算值Table 3 Experimental and calculated vibrational frequencies for free and adsorbed methanol
3.1.3 红外振动频率分析
本文还计算了甲醇分子吸附前后的振动频率,并与实验值39一起列于表3.由表可见,吸附后C―O和O―H键振动频率降低,发生红移,尤其是νO―H降低最多,从3723红移至3337 cm-1,说明O―H键活化程度高.由此可预测,甲醇分子的解离途径是断裂O―H键,生成甲氧基(CH3O―)中间体.
3.2 甲氧基在FeS2(100)表面的吸附
3.2.1 吸附能与吸附构型
同样考虑甲氧基分别以C―O键垂直和平行方式吸附于FeS2(100)表面的四个吸附位,计算后发现CH3O―吸附后都转化为同一种构型.即甲氧基以C―O键倾斜吸附于表面,通过O原子与表面Fe成键,构型如图4所示.吸附能Eads=151.5 kJ·mol-1,可见甲氧基与底物发生较强的相互作用.
具体构型参数和实验值40列于表4,由于吸附后转化为同一种构型,表中只列出Vert-Fe-top构型参数.由表可见C―O键键长伸长比较明显,由0.1344 nm伸长到0.1394 nm,C―H键键长基本没有变化,角度θCOH有所扩大.O原子与Fe2+形成的O―Fe键长为0.1883 nm,小于甲醇与底物形成的O―Fe键长0.2122 nm,再次表明了甲氧基与底物的相互作用较强.
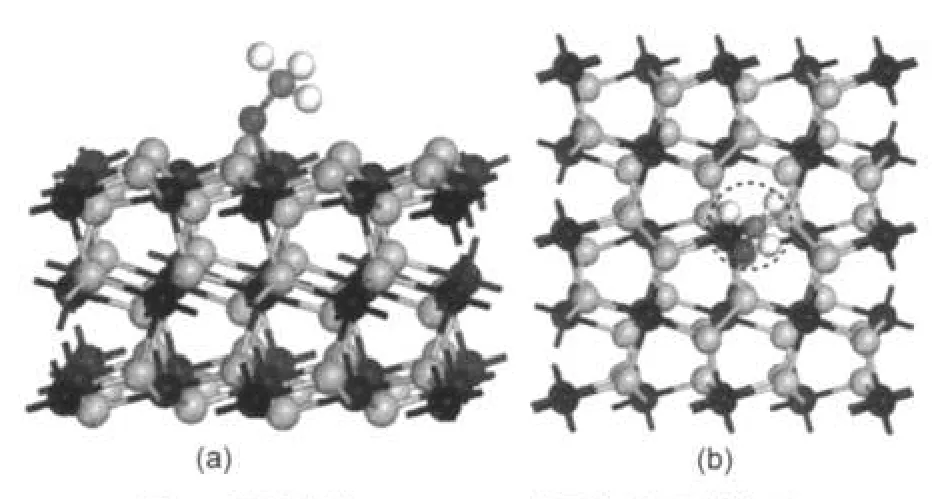
图4 甲氧基在FeS2(100)表面稳定吸附构型的侧视图(a)和俯视图(b)Fig.4 Side view(a)and top view(b)of stable structure of CH3O―adsorbed on FeS2(100)surface

表4 甲氧基在FeS2(100)面吸附的吸附能和几何构型参数Table 4 Adsorption energy and geometrical parameters for methoxy adsorbed on FeS2(100)surface
3.2.2 Mulliken电荷布居和轨道分析
表5给出了甲氧基各原子和Fe2+的轨道电荷布居,由表可见,甲氧基吸附后带负电荷,体系的电子由底物转移到吸附质.在吸附过程中,O原子与Fe2+成键,Fe2+3d电子转移到电负性较强的O原子的2p轨道,因此吸附后甲氧基得电子.由吸附体系的分子轨道图也可以看出,甲氧基主要通过氧原子与底物作用,图5给出了甲氧基自由态和吸附后体系的前线分子轨道.由图可见,甲氧基的HOMO是n轨道,主要成分来自于O原子的孤对电子.在吸附体系中,O原子的2p轨道与Fe2+的3d轨道相互作用成键,Mulliken电荷布居分析与轨道分析结果一致.
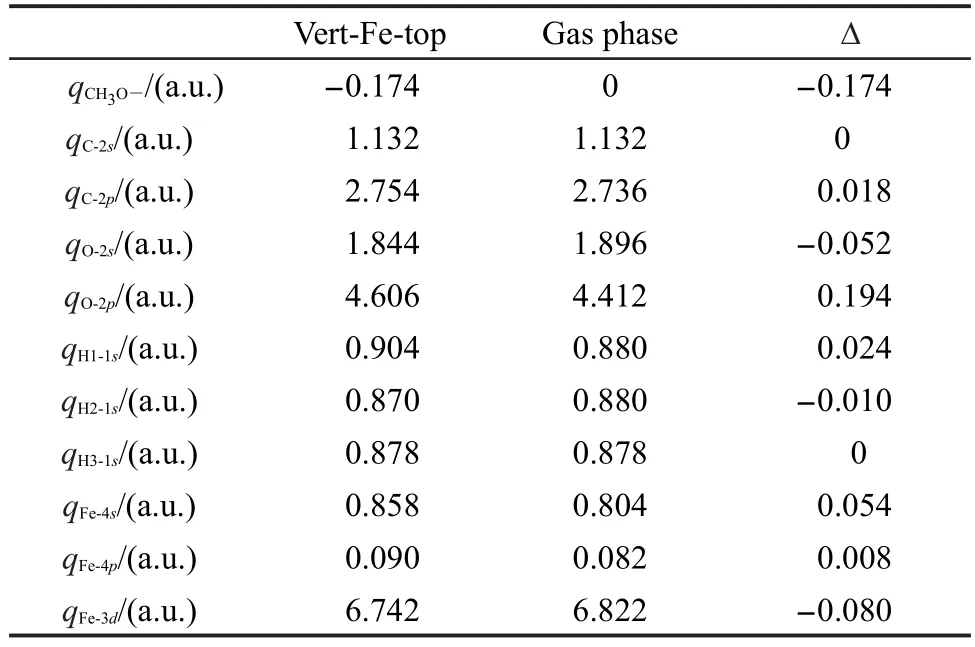
表5 甲氧基各原子和Fe2+的轨道电荷布居Table 5 Orbital populations for atoms of methoxy and Fe2+before and after adsorption
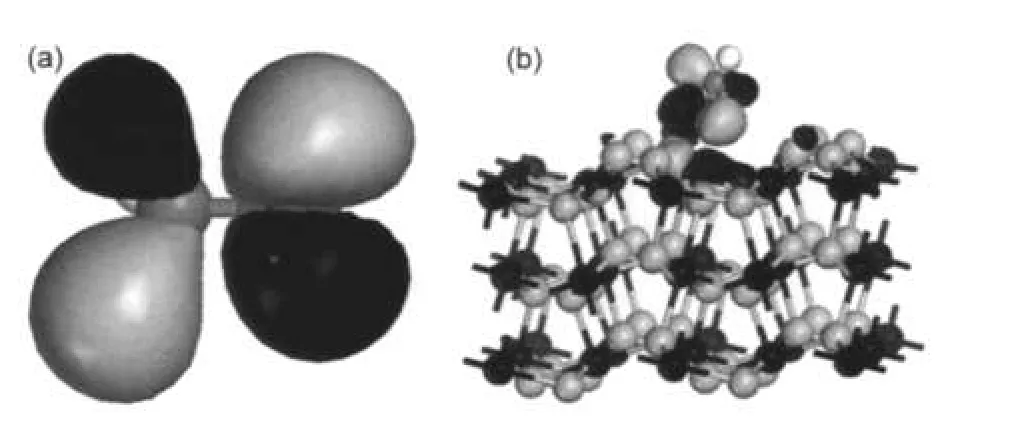
图5 自由甲氧基(a)和吸附后吸附体系(b)的HOMO图Fig.5 HOMOs of free CH3O―(a)and the adsorption system(b)
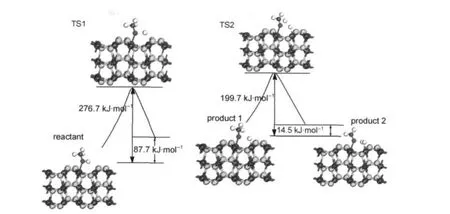
图6 甲醇在FeS2(100)表面的可能的解离途径示意图Fig.6 Possible decomposition pathway of methanol on FeS2(100)surface
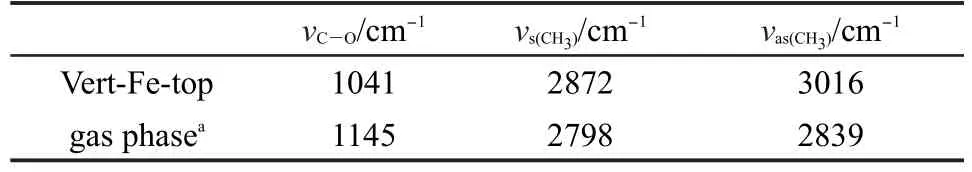
表6 甲氧基吸附前后的振动频率Table 6 Vibrational frequencies for free and adsorbed methoxy
3.2.3 红外振动频率分析
本文还计算了甲氧基吸附前后的振动频率,由表6可见,吸附后C―O键振动频率降低,发生红移,从1145 cm-1红移至1041 cm-1,说明CH3O得电子, C―O键得到活化,与前面结果分析一致.CH3―的对称反对称伸缩振动频率提高,发生蓝移.
3.3 甲醇在FeS2(100)表面的解离吸附
本文还探讨了甲醇分子在FeS2(100)面可能的解离途径,通过前面的讨论可知,甲醇以C―O键倾斜吸附于底物的Fe-top位,甲氧基也是如此.我们还计算了H原子在Fe(100)表面的吸附,结果是表面Fe位是H原子的稳定吸附位,吸附能为197.7 kJ· mol-1.根据文献报道,18,41我们认为甲醇分子解离的途径可能为:首先,甲醇分子断裂羟基的O―H键,生成甲氧基和H原子分别吸附于底物表面的两个Fe2+;其次,甲氧基断裂一个C―H键,生成甲醛和H原子,而两个H原子结合成氢气吸附于Fe2+.计算采用complete LST/QST的方法寻找过渡态,计算方法以及其他参数均与前面构型优化时相同.解离的起点(反应物reactant)取甲醇吸附后的稳定构型Vert-Fe-top,经历过渡态(TS1),得到第一步解离产物甲氧基和H原子(product 1);进一步搜索第二步的过渡态(TS2),得到主要产物HCHO和H2(product 2),如图6所示.
在解离吸附过程中,甲醇的羟基H―O键先断裂,生成产物甲氧基和氢原子.搜索到过渡态中的氢氧之间的距离由分子时的0.0989 nm增加到0.2017 nm,随着羟基的断裂以及Fe―H键和Fe―O键的形成,该反应能垒为276.7 kJ·mol-1,小于在气相中断裂羟基所需要的能量(430 kJ·mol-1).由此说明在FeS2(100)表面甲醇更容易解离,FeS2对甲醇的解离起到了一定的催化作用.该反应属于吸热反应,反应热需要87.7 kJ·mol-1.形成的Fe―O键键长为0.1879 nm,比Fe原子与O原子的半径和0.1940 nm短,说明甲氧基的O原子与Fe2+之间产生了强烈的化学作用.而Fe―H键键长为0.1521 nm,与Fe原子和H原子的半径和0.1490 nm非常接近,表明H与Fe2+之间也形成了化学键,两者的相互作用也较强.
甲醇进一步解离,最后产物为HCHO和H2.该解离反应的能垒为199.7 kJ·mol-1,反应需要吸热14.5 kJ·mol-1.说明O―H键的断裂需要较大的能量,由此可见甲醇在FeS2表面解离中第一步解离为速控步骤,从而为理解煤矿中FeS2的分离及抑制FeS2的氧化提供理论基础..
4 结论
本文采用密度泛函理论结合周期平板模型的方法,计算了甲醇在(2×2)超晶胞九层平板模型FeS2(100)面的吸附,并探究了甲醇可能的解离途径.甲醇以及解离产物甲氧基、氢原子和甲醛均倾向于吸附在Fe-top位,甲醇与甲氧基的吸附能分别为69.3和151.5 kJ·mol-1.在吸附过程中,甲醇分子的构型发生变化,C―O键与O―H键的键长均有不同程度的伸长,C―H键的键长变化较小.
Mulliken电荷布居分析揭示吸附过程中电子由甲醇转移到FeS2(100)面,而红外振动频率的计算表明C―O和O―H键振动频率发生红移,特别是O―H的活化程度更高.过渡态的计算发现甲醇在FeS2(100)表面解离过程中,生成甲氧基中间体时需要反应能垒较高,为反应的决速步骤,并最终生成甲醛和氢气,从而为理解FeS2(100)表面与界面提供理论基础.
(1) Brown,J.R.;Bancroft,G.M.;Fyfe,W.S.;Mclean,R.A.N. Environ.Sci.Technol.1979,13(9),1142.
(2)Wang,D.Z.;Long,X.Y.;Sun,S.Y.Chin.J.Nonfer.Metals 1991,1(1):15.[王淀佐,龙翔云,孙水裕.中国有色金属学报,1991,1(1),15.]
(3) Lalvani,S.B.;DeNeve,B.A.;Weston,A.Corrosion Sci.1991, 47(1),55.
(4) Ogunsola,O.M.;Osseo-Assare,K.Fuel 1987,66(4),467.
(5) Olson,T.J.;Aplan,F.F.Processing and Utilization of High Sulfur Coal;Chug,Y.P.,Cauldle,R.D.Eds.;Elsevier: Amsterdam,1987;p 71.
(6) Singer,P.C.;Stumm,W.Science 1970,167(1),1121.
(7) Lowson,R.T.Chem.Rev.1982,82(5),461.
(8) Nordstrom,D.K.SSSA Special Publication 1982,10(5),37.
(9) Huang,X.;Evangelou,V.P.Environmental Geochemistry of Sulfide Oxidation,ACS Symp.Ser.550,1994;Alpers,C.N., Blowes,D.W.Eds.;Oxford University Press:New York; Chapter 34,p 562.
(10) Ennaoui,A.;Fiechter,S.;Jaegermann,W.;Tributsch,H. J.Electrochem.Soc.1986,133(1),97.
(11) Nesbitt1,H.W.;Scaini,M.;Höchst,H.;Bancroft,G.M.; Schaufuss,A.G.;Szargan,R.Am.Mineral.2000,85(5-6),850.
(12) Uhlig,I.;Szargan,R.;Nesbitt,H.W.;Laajalehto,K.Appl.Surf. Sci.2001,179,222.
(13) Descostes,M.;Mercier,F.;Beaucaire,C.;Zuddas,P.;Trocellier, P.Nucl.Instru.Meth Phys.Res.B 2001,181(1-4),603.
(14) Mattila,S.;Leiro,J.A.;Laajalehto,K.Appl.Surf.Sci.2003, 212,97.
(15) Leiro,J.A.;Mattila,S.S.;Laajalehto,K.Surf.Sci.2003,547 (1-2),157.
(16) Mattila,S.;Leiro,J.A.;Heinonen,M.Surf.Sci.2004,566-568, 1097.
(17) Kim,E.J.;Batchelor,B.Environ.Sci.Technol.2009,43(8), 2899.
(18) Guevremont,J.M.;Strongin,D.R.M.;Schoonen,A.A.Surf. Sci.1997,391(1-3),109.
(19) Stirling,A.;Bernasconi,M.;Parrinello,M.J.Chem.Phys. 2003,118(19),8917.
(20) Stirling,A.;Bernasconi,M.;Parrinello,M.J.Chem.Phys. 2003,119(9),4934.
(21) Boehme,C.;Marx,D.J.Am.Chem.Soc.2003,125(44),13362.
(22)Sun,W.;Hu,Y.H.;Qiu,G.Z.;Qin,W.Q.J.Cent.South Univ. Technol.2004,11(4),386. [孙 伟,胡岳华,邱冠周,覃文庆.中南工业大学学报,2004,11(4),386.]
(23)Von Oertzen,G.U.;Skinner,W.M.;Nesbitt,H.W.Phys.Rev.B 2005,72(23),235427.
(24) Nair,N.N.;Schreiner,E.;Marx,D.J.Am.Chem.Soc.2006, 128(42),13815.
(25) Li,Q.;Qin,W.Q.;Sun,W.;Qiu,G.Z.J.Cent.South Univ. Technol.2007,14(5),618.[黎 全,覃文庆,孙 伟,邱冠周.中南工业大学学报,2007,14(5),618.]
(26) Blanchard,M.;Wright,K.;Gale,J.D.;Catlow,C.R.A.J.Phys. Chem.C 2007,111(30),11390.
(27) Kleppe,A.K.;Jephcoat,A.P.Mineralogical Magazine 2004,68 (3),433.
(28)Du,Y.D.;Chen,W.K.;Zhang,Y.F.;Guo,X.J.Nat.Gas Chem. 2011,20(1),60.
(29) Parr,R.G.;Yang,W.Density Functional Theory of Atoms and Molecules;Oxford University Press:New York,1989;p 1.
(30) Cao,M.J.;Chen,W.K.;Liu,S.H.;Xu,Y.;Li,J.Q.Acta Phys.-Chim.Sin.2006,22(1),11. [曹梅娟,陈文凯,刘书红,许 莹,李俊籛.物理化学学报,2006,22(1),11.]
(31) Delley,B.J.Chem.Phys.1990,92(1),508.
(32) Delley,B.J.Chem.Phys.2000,113(18),7756.
(33) Lide,D.R.CRC Handbook of Chemistry and Physics.84th ed; CRC Press:Boca Raton,2003-2004;pp 9-34.
(34) Redhead,P.A.Vacuum 1962,12(4),203.
(35)Chen,W.K.;Liu,S.H.;Cao,M.J.;Lu,C.H.;Xu,Y.;Li,J.Q. Chin.J.Chem.2006,24(7),872.
(36)Wang,Y.W.;Li,L.C.;Tian,A.M.Acta Chim.Sin.2008,66 (22),2457.[王译伟,李来才,田安民.化学学报,2008,66 (22),2457.]
(37) Jiang,S.Y.;Teng,B.T.;Lu,J.Q.;Liu,X.S.;Yang,P.F.;Yang, F.Y.;Luo,M.F.Acta Phys.-Chim.Sin.2008,24(11),2025. [蒋仕宇,滕波涛,鲁继青,刘雪松,杨培芳,杨飞勇,罗孟飞.物理化学学报,2008,24(11),2025.]
(38) Rosso,K.M.;Becker,U.;Hochella,M.F.Am.Mineral.1999, 84(10),1535.
(39) Herzberg,G.Molecular Spectra and Molecular Structure ІІ. Infrared and Raman Spectra of Polyatomic Molecules;D.Van Nostrand Company:New York,1945;p 335.
(40) Jackels,C.F.J.Chem.Phys.1982,76(1),505.
(41) Lu,J.P.;Albert,M.;Bernasek,S.L.Surf.Sci.1990,239(1-2), 49.
December 15,2010;Revised:March 6,2011;Published on Web:March 24,2011.
Adsorption and Dissociation of Methanol on Perfect FeS2(100)Surface
DU Yu-Dong1ZHAO Wei-Na1GUO Xin2ZHANG Yong-Fan1CHEN Wen-Kai1,*
(1Department of Chemistry,Fuzhou University,Fuzhou 350108,P.R.China;
2State Key Laboratory of Coal Combustion,Huazhong University of Science and Technology,Wuhan 410074,P.R.China)
First-principles calculations based on density functional theory(DFT)and the periodical slab model were used to study the adsorption and dissociation of methanol on the perfect FeS2(100)surface. The adsorption energy and the geometric parameters on the different adsorption sites showed that the Fe site was the most favorable adsorption site and O atoms were found to bind to Fe atoms.After adsorption, the C―O and O―H bonds of methanol were elongated and the vibrational stretch frequency was red shifted.The calculation results proved that methanol was prone to decomposition resulting in methoxy groups and hydrogen.We calculated the adsorption behavior of these methoxy groups and hydrogen on the FeS2(100)surface and found that the Fe sites were also the most favorable adsorption sites.A possible decomposition pathway was investigated using transition state searching methods:first the O―H bond of methanol was decomposed producing the intermediate methoxy group and subsequently the C―H bond of the methoxy group was broken resulting in final products of formaldehyde and hydrogen.
Density functional theory;Methanol;FeS2(100)surface;Adsorption;Transition state
O641
∗Corresponding author.Email:qc2008@fzu.edu.cn;Tel:+86-591-22866162.
The project was supported by the National Natural Science Foundation of China(90922022),State Key Laboratory of Coal Combustion Foundation of Huazhong University of Science and Technology,China(FSKLCC0814),and New Century Excellent Talents Program in University of Fujian Province,China(HX2006-103).
国家自然科学基金(90922022),华中科技大学煤燃烧国家重点实验室基金(FSKLCC0814)和福建省高等学校新世纪优秀人才计划(HX2006-103)资助项目
猜你喜欢
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
科学(2020年2期)2020-08-24
山东化工(2019年18期)2019-10-16
铜仁学院学报(2018年6期)2018-07-05
科技资讯(2017年12期)2017-06-09
中成药(2017年4期)2017-05-17
中成药(2017年3期)2017-05-17
北京航空航天大学学报(2017年10期)2017-04-20
航天返回与遥感(2014年4期)2014-07-31
无机化学学报(2014年4期)2014-02-28
