
UPLC-MS-MS法测定水产品中克百威及其代谢物
2012-12-27 08:50何亚斌
食品与机械 2012年4期
何亚斌
(农业部食品质量监督检验测试中心,上海 200436)
UPLC-MS-MS法测定水产品中克百威及其代谢物
何亚斌
(农业部食品质量监督检验测试中心,上海 200436)
采用超高效液相色谱-串联质谱技术建立同时测定水产品中克百威及其代谢物(3-羟基克百威、3-酮基克百威和呋喃酚)含量的方法。样品经乙腈提取,正己烷脱脂,HLB固相萃取柱净化,色谱柱分离,电喷雾串联四极杆质谱进行检测,采用多反应监测分析,并对液质分离条件及参数和样品前处理条件进行优化。结果显示,克百威及其代谢物在0.08~100ng/mL范围内线性良好(0.998 3~0.999 7)。在0.25~2.5μg/kg时,平均加标回收率在87.9%~96.1%,RSD值在1.4%~9.5%。该方法测定克百威、3-羟基克百威、3-酮基克百威和呋喃酚的检出限均为0.25μg/kg。该方法快速、准确、灵敏,可用于水产品中克百威及其代谢物残留量的测定。
超高效液相色谱-串联质谱;水产品;克百威;3-羟基克百威;3-酮基克百威;呋喃酚
克百威是一种广谱内吸杀虫杀螨剂,可用于多种作物防治土壤内及地面上的300多种害虫和线虫[1,2]。克百威对胆碱酯酶抑制的不可逆性决定了其对人、畜、禽、鱼等的毒性极高[3-5],目前,克百威在种植业中已经禁止使用。但是克百威在土壤中的残留期较长,在土壤中的移动性能较大(水溶解度为700mg/L),在降水量大、地下水位浅的砂土地区易引起对地下水的污染[6]。同时,鱼体对呋喃丹具有一定的富集,而且残留浓度较高[7]。目前在中国农业部第235号公告中规定动物性食品中不得检出克百威。在GB 11607——1989中规定克百威的标准值要≤0.01mg/L,但是在中国缺乏水产品中克百威的检测方法标准。克百威在碱性介质下分解较快,可降解为3-羟基克百威、3-酮基克百威,最终降解成相应的酚类[8]。因此,测定克百威的残留量,除测定克百威含量外,还需测定3-羟基克百威、3-酮基克百威和呋喃酚等代谢物。
目前报道的关于测定克百威的方法很多[9-11],但未见同时测定水产品中克百威及其多种代谢物的报道。本试验拟采用UPLC-MS-MS法,建立同时测定水产品中克百威及其代谢物(3-羟基克百威、3-酮基克百威、呋喃酚)的方法。
1 材料与方法
1.1 材料与试剂
水产品:鱼、虾,购自上海曹安农贸市场;
乙腈、甲酸和正己烷:色谱纯,美国Sigma公司;
氯化钠:分析纯,国药集团化学试剂有限公司;
标准品:克百威、3-羟基克百威、3-酮基克百威、呋喃酚,德国Dr.Ehrenstorfer公司;
Oasis HLB固相萃取柱:60mg/3mL,美国沃特世公司。
1.2 仪器与设备
超高效液相色谱系统:ACQUITY UPLC,美国沃特世公司;
串联四极杆质谱仪(配有电喷雾离子源(ESI)及 Masslynx 4.0数据工作站):Waters XEVO TQ,美国沃特世公司;
离心机:Heraeus Multifuge X1R,美国热电公司;
氮吹仪:ANPEL DC12,上海安谱科学仪器有限公司;
超声波提取仪:EDAA-2500TH,上海安谱科学仪器有限公司;
固相萃取装置:SBAB-57030U,上海安谱科学仪器有限公司。
1.3 样品前处理
1.3.1 提取 取500g代表性水产品样品可食性部分,粉碎均匀,冷冻保存。准确称取5.00g样品,于50mL离心管中,加入3~5g氯化钠,加入乙腈15mL,均质2min,4℃,5 000r/min离心5min,取上清液于50mL离心管中。再用15mL乙腈分两次重复以上步骤提取残渣,合并上清液。在提取液中加入10mL乙腈饱和过的正己烷,剧烈振荡,于4℃,5 000r/min离心3min,弃去正己烷相,再用10mL乙腈饱和过的正己烷,重复以上步骤。有机相转入鸡心瓶中,40℃水浴旋转蒸干。加入30%乙腈水溶液1.0mL溶解残渣,再加入9.0mL蒸馏水,混合均匀,供SPE柱净化。
1.3.2 净化 将全部样品提取液过Oasis HLB SPE柱 (预先用3mL甲醇、3mL水活化),上样后再用6mL水淋洗,淋洗后在固相萃取装置上真空抽干水分10min,经6mL甲醇溶液洗脱,收集至10mL玻璃试管中,50℃氮气吹至近干,加入1mL流动相,充分旋涡混合,经0.22μm滤膜过滤,待UPLC-MS-MS分析。
1.4 仪器条件
1.4.1 色谱条件 色谱柱:ACQUITY HSS T3(1.8μm,2.1mm ×100mm),流动相:乙腈(0.1%甲酸)-5mmol/L乙酸铵水溶液(0.1%甲酸),梯度洗脱程序见表1。流速0.3mL/min,柱温40℃,进样量5μL。
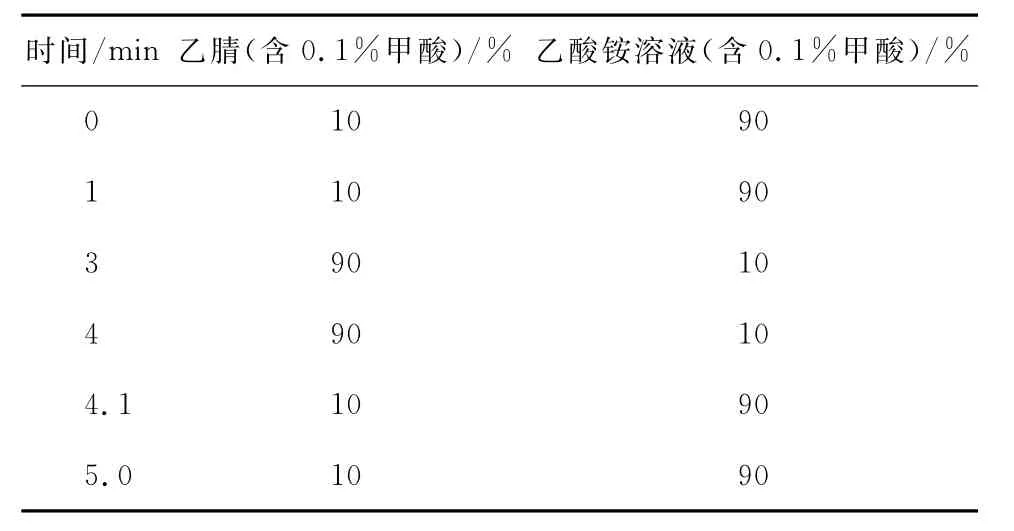
表1 流动相梯度洗脱程序Table 1 Gradient elution program
1.4.2 质谱条件 电喷雾离子源,正离子扫描模式,多反应监测MRM,毛细管电压1.5kV,离子源温度150℃,雾化温度400℃,雾化气流速800L/h,二级碰撞气为氩气,碰撞气流速0.15mL/min,其他质谱参数见表2。
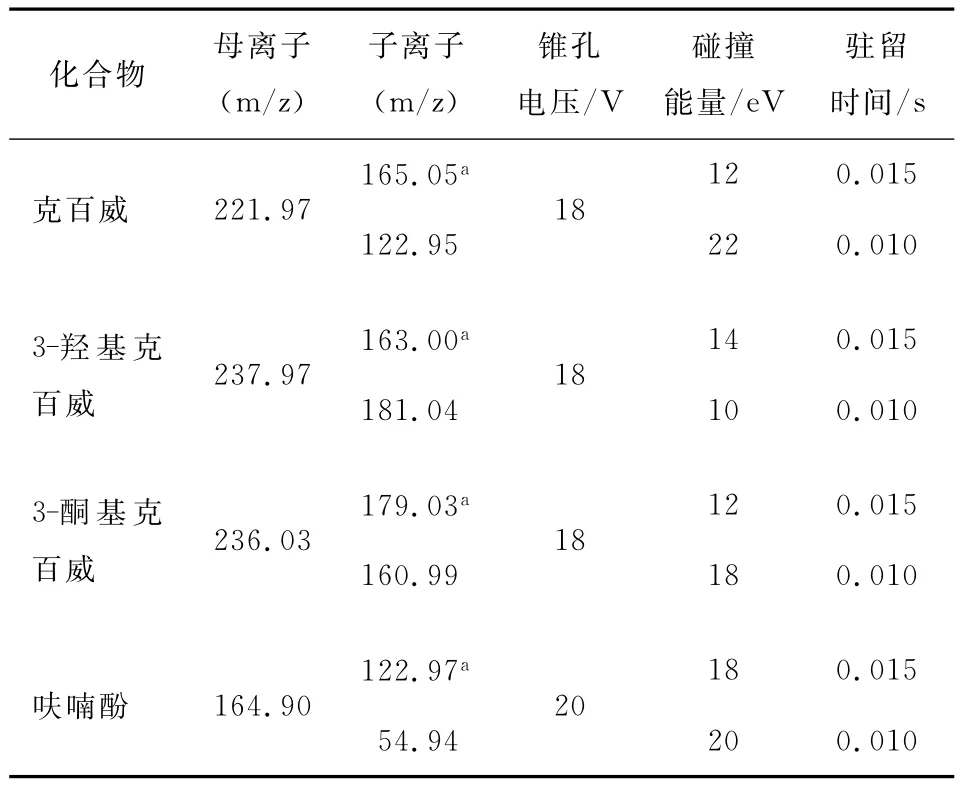
表2 主要参考质谱参数ńTable 2 MS/MS parameters
2 结果与讨论
2.1 提取条件的选择
乙腈作为一种通用的提取溶剂在残留检测方面得到广泛的应用,由于其对脂肪、蛋白质类化合物较难提取,对绝大多数农药有较高的回收率。因此,本试验采用乙腈作为提取试剂,再使用乙腈饱和的正己烷去除脂肪,在尽量少地带入脂肪、蛋白类化合物的前提下,最大限度的提取了水产品中克百威及其代谢物。
2.2 净化条件的选择
本试验分别考察了HLB、C18两种填料的固相萃取小柱的净化效果。试验发现,克百威及其代谢物在HLB、C18小柱具有一定的保留,通过水直接淋洗去除有机酸等水溶性杂质,再用甲醇洗脱目标化合物。HLB小柱对克百威及其代谢物柱效回收率都大于95%,但C18小柱对3-羟基克百威的保留效果不好,柱效回收率都只有60%左右。因此,采用HLB小柱净化。
HLB小柱对克百威、3-酮基克百威和呋喃酚有很好的保留,但是,3-羟基克百威在HLB小柱上的保留比较弱。试验结果表明:50%的乙腈水溶液不会将吸附在HLB小柱上的克百威、3-酮基克百威和呋喃酚被洗脱下来,乙腈含量达到90%以上才能逐步洗脱下来;5mL 10%乙腈水溶液洗脱就可以将吸附在HLB小柱上的3-羟基克百威洗脱68%,而即使10mL 3%乙腈水溶液淋洗也不会将3-羟基克百威洗脱下来。试验中发现30%乙腈水溶液能全部溶解残渣中的4种化合物。因此,本方法中在乙腈提取浓缩后,用1.0mL 30%乙腈水溶液溶解残渣,再加入9.0mL纯水稀释之后上净化柱。
2.3 仪器分析条件的选择
本试验采用乙腈(0.1%甲酸)-5mmol/L乙酸铵水溶液(0.1%甲酸)作为流动相;流动相中加入0.1%甲酸增加了目标化合物在喷雾前的离子化程度,提高了检测的灵敏度;同时加入5mmoL乙酸铵,改善了目标化合物的峰形,离子流图见图1~4。本试验采用HSS T3色谱柱,利用其对极性化合物强保留的特点,实现了对克百威、3-羟基克百威、3-酮基克百威和呋喃酚的同时测定。在本试验采用的流动相、色谱柱和洗脱比例条件下,克百威和呋喃酚未能有效分离,但由于本试验采用多反应检测的扫描方式,对目标化合物的定性和定量都不影响。
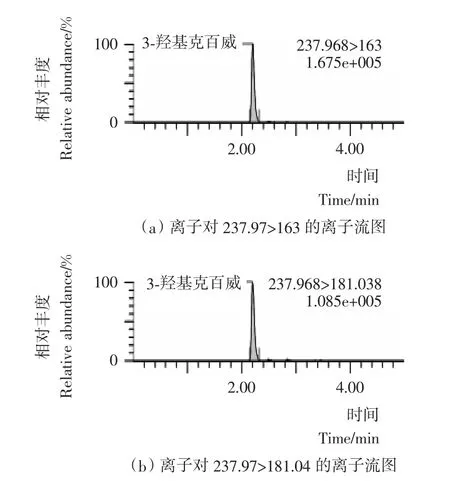
图1 3-羟基克百威标准物质选择离子流图Figure 1 Selected ion chromatograms of 3-hydroxy carbofuran
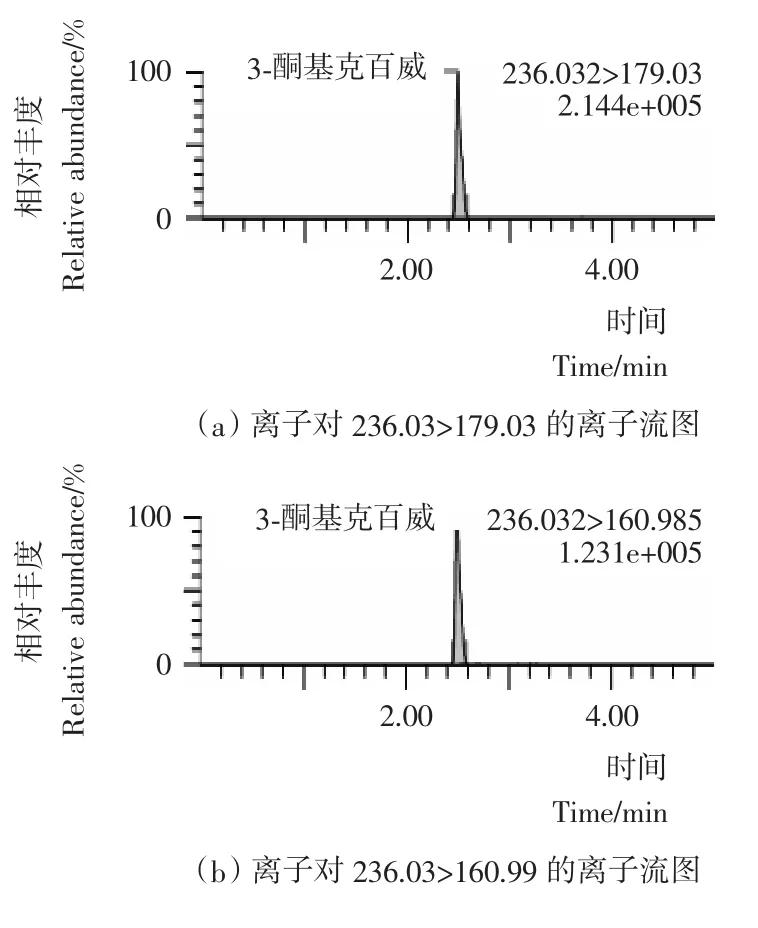
图2 3-酮基克百威标准物质选择离子流图Figure 2 Selected ion chromatograms of 3-keto carbofuran
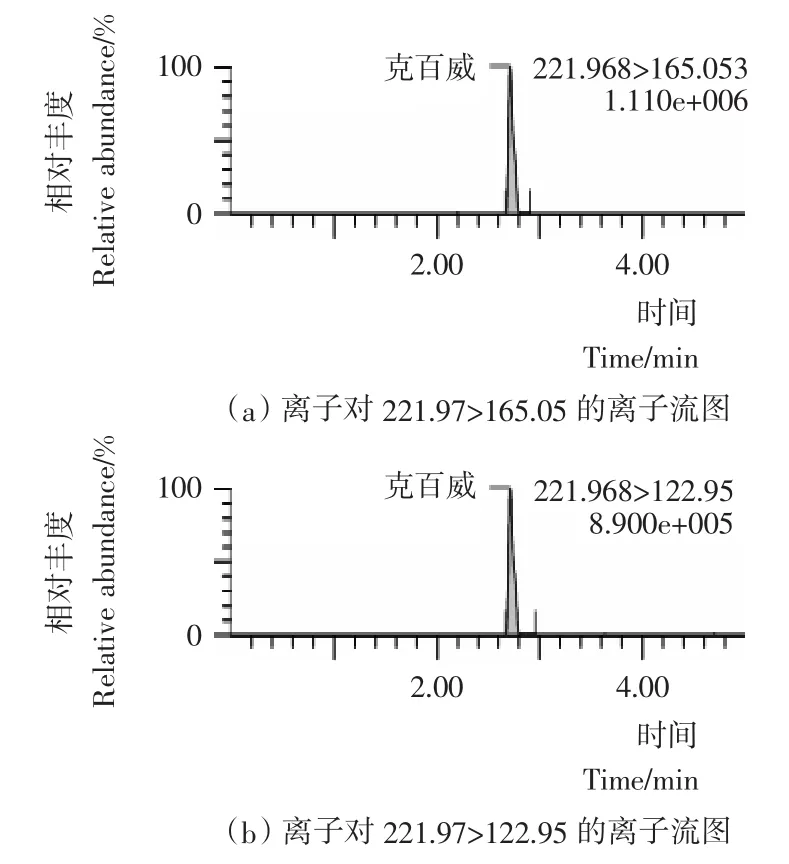
图3 克百威标准物质选择离子流图Figure 3 Selected ion chromatograms of carbofuran
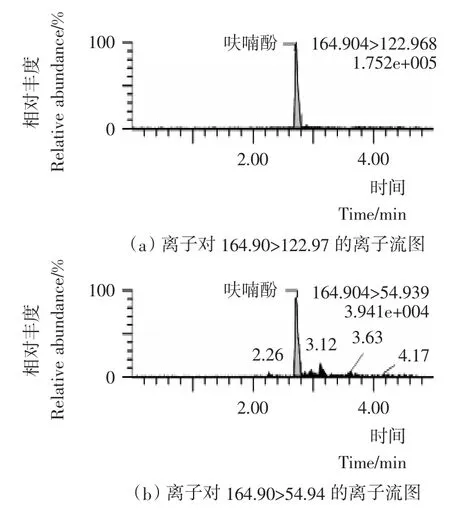
图4 呋喃酚标准物质选择离子流图Figure 4 Selected ion chromatograms of carbofuran phenol
将克百威、3-羟基克百威、3-酮基克百威和呋喃酚标准品溶液采用流动注射直接进样,通过全扫描确定化合物母离子,各化合物均选用[M+H]+作为母离子,再对母离子进行二级质谱扫描,得到碎片离子,通过优化条件,得到二级质谱图。通过多反应离子监测(MRM)选择相对丰度较高的离子对,确定为定性定量离子对,并优化毛细管电压、锥孔电压、碰撞电压等参数,优化后的质谱条件见表2。
2.4 线性及灵敏度
以空白样品基质配制一系列不同浓度的混合标准工作溶液(0.08,0.16,0.80,4.00,20.00,50.00,100.00ng/mL),并依次进样,分别以克百威、3-羟基克百威、3-酮基克百威和呋喃酚的峰面积y为纵坐标,以相应的浓度值x为横坐标,作标准曲线,结果表明,这4种化合物在0.08~100ng/mL范围内其浓度与峰面积呈良好的线性关系,相关系数r大于0.998,见表3。
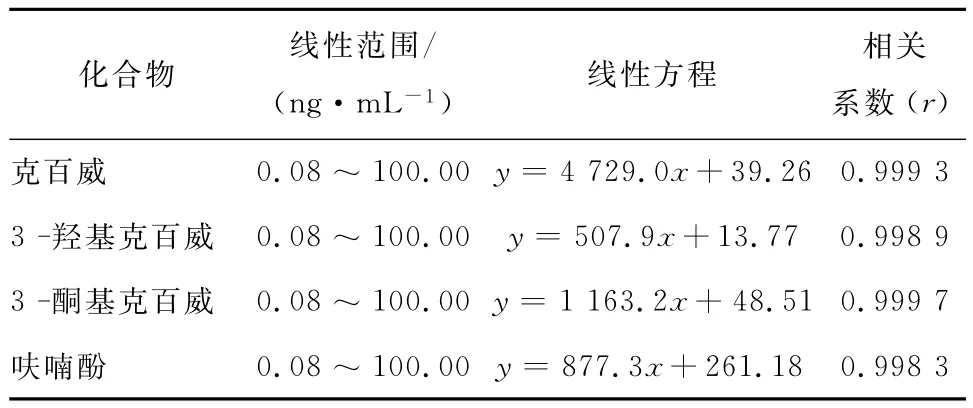
表3 克百威、3-羟基克百威、3-酮基克百威和呋喃酚的线性方程及相关系Table 3 Linear equations and correlation coefficients of four target compounds
在水产品样品中添加最低检出限相当浓度的一系列浓度的标样,由基质空白所产生的仪器背景信号的3倍值的相应量来验证方法的灵敏度。克百威、3-羟基克百威、3-酮基克百威和呋喃酚的最低检测限(LOD)为0.25μg/kg。
2.5 准确度和精密度
采用标准添加法,在5.00g阴性样品中进行3个不同水平(0.25,0.50,2.50μg/kg)的添加回收试验,测定后得到克百威、3-羟基克百威、3-酮基克百威和呋喃酚的回收率和相对标准偏差(RSD)见表4。由表4可知,在鱼肉和虾仁样品中3个添加水平上,4种化合物的平均回收率在87.9%~96.1%,RSD 在1.4%~9.5%,符合农药多残留检测的要求[12]。
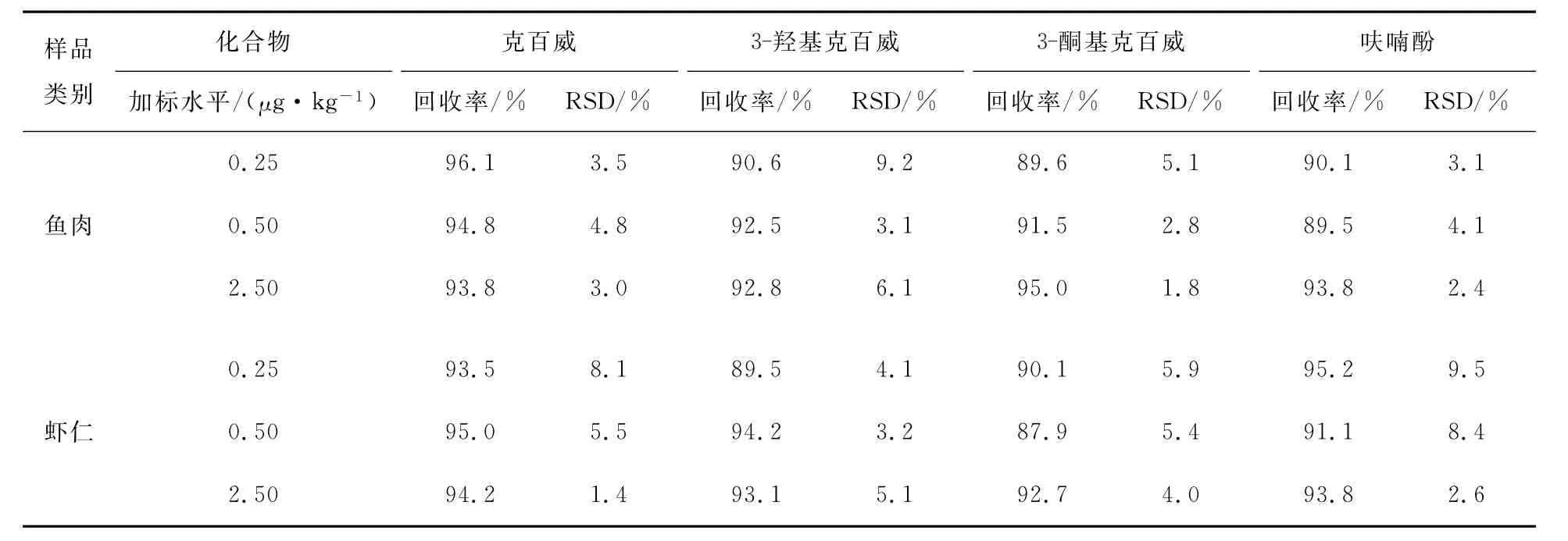
表4 鱼肉、虾仁中克百威、3-羟基克百威、3-酮基克百威和呋喃酚的回收率和相对标准偏差Table 4 Spiked recoveries and RSDs of four target compounds in fish and shrimp(n= 6)
3 结论
本试验建立了水产品中克百威及其代谢物的超高效液相色谱-串联质谱(UPLC-MS-MS)检测方法。采用乙腈提取,正己烷去除脂肪,HLB固相萃取小柱净化,消除了水产品中蛋白、有机酸及脂类物质的干扰,再应用ACQUITY HSS T3色谱柱分离,电喷雾串联四极杆质谱进行检测,采用多反应监测(MRM)分析行色谱分离,实现了对克百威及其代谢物定性、定量测定。该检测方法快速、灵敏、可靠。
1 唐除痴,李煜超,陈彬,等.农药化学[M].天津:南开大学出版社,1997:169~170.
2 陈建波,王云飞,奚道珍,等.克百威及其代谢产物残留分析方法研究进展[J].农药科学与管理,2011,32(3):31~33.
3 徐赫.呋喃丹致畸性研究[J].环境科学研究,1989,2(1):27~29.
4 Ferguson P W,Eey M S,Jewell S A,et al.Carbofuran metabolism and toxicity in the rat[J].Fundamental and Applied Toxi-cology,1984,4(1):14~21.
5 Winston H H.Public health goal for chemicals in drinking water:carbofuran[R].California:California Environmental Protection Agency,2000.
6 李娟,赵永刚,丁曦宁.固相萃取/高效液相色谱法测定地表水中氨基甲酸醋类农药[J].环境监测管理与技术,2006,18(1):27~28.
7 王森,李荣,邹世平.气相色谱法测定水产品中呋喃丹的残留量[J].色谱,2008,26(6):775~777.
8 卢培标,戴维列.呋喃丹及其主要水解、代谢产物的检验[J].分析测试学报,1998,17(5):81~83.
9 符展明,金米聪,金永高,等.GC/MS法测定蔬菜中克百威残留量的研究[J].中国卫生检验杂志,2005,15(4):421~422.
10 谭头云,蔡磊明,贾福艳,等.气相色谱法测定大豆中克百威及其代谢物的残留量[J].农药,2009,48(1):58~59.
11 陈笑梅,胡贝贝,刘海山.高效液相色谱-串联质谱法测定粮谷中9种氨基甲酸酯类农药残留[J].分析化学,2007,35(1):106~110.
12 刘光学,乔雄梧,陶传江,等.NY/T 788——2004农药残留试验准则[S].北京:中国农业出版社,2004.
Determination of carbofuran and its metabolites in aquatic products by UPLC-MS-MS
HE Ya-bin
(Food Quality Supervision and Inspection Center of China Ministry of Agriculture,Shanghai200436,China)
An ultra performance liquid chromatography tandem mass spectrometric method(UPLC-MS/MS)has been developed for the simultaneous determination of carbofuran and its main metabolites(3-hydroxycarbofuran,3-ketocarbofuran and carbofuran phenol)in aquatic products.The carbofuran and its main metabolites were extracted with acetonitrile,cleaned up with HLB solid phase extraction cartridge(SPE).The UPLC analyses were performed on an ACQUITY HSS T3column with gradient evaluation,combined with electrospray ionization in positive mode(ESI+)and multiple reaction monitoring(MRM)mode.The linear range was 0.08~100ng/mL,and the correlation coefficients were 0.998 3~0.999 7.The average recoveries of carbofuran and its main metabolites in fish and shrimp(0.25to 2.5μg/kg)ranged from 87.9%to 96.1%and the relative standard deviations were between 1.4%and 9.5%.The detection limit of carbofuran,3-hydroxy carbofuran,3-keto carbofuran and carbofuran phenol was 0.25μg/kg.The method is easy,rapid,sensitive and suitable for the determination of carbofuran and its main metabolites in aquatic products.
Ultra performance liquid chromatography-tandem mass spectrometry(UPLC- MS- MS);aquatic products;carbofuran;3-hydroxycarbofuran;3-ketocarbofuran;carbofuran phenol
10.3969 /j.issn.1003-5788.2012.04.024
上海市技术型贸易措施应对专项(编号:10TBT004)
何亚斌(1980-),男,农业部食品质量监督检验测试中心(上海)工程师,硕士。E-mail:heyabin@shdenuo.com
2012-05-12
猜你喜欢
少年文艺(2022年8期)2022-07-08
科学与财富(2021年34期)2021-05-10
浙江农业学报(2020年11期)2020-12-02
中国经济周刊(2017年6期)2017-03-21
烟草科技(2015年8期)2015-12-20
中国医疗美容(2015年1期)2015-07-12
无机化学学报(2014年6期)2014-02-28
食品科学(2013年24期)2013-03-11
食品科学(2013年19期)2013-03-11
故事林(2010年23期)2010-05-14
