
Origin of the cis-Effect:a Density Functional Theory Study of Doubly Substituted Ethylenes
2013-07-25 09:12ZHAODongBoRONGChunYingJENKINSSamanthaKIRKStevenYINDuLinLIUShuBin
物理化学学报 2013年1期
ZHAO Dong-Bo RONG Chun-Ying,* JENKINS Samantha KIRK Steven R.,*YIN Du-Lin LIU Shu-Bin,2,*
(1Key Laboratory of Chemical Biology and Traditional Chinese Medicine Research(Ministry of Education of China)and Key Laboratory of Resource Fine-Processing and Advanced Materials of Hunan Province,College of Chemistry and Chemical Engineering,Hunan Normal University,Changsha 410081,P.R.China;2Research Computing Center,University of North Carolina,Chapel Hill,North Carolina 27599-3420,U.S.A.)
1 lntroduction
It is common sense that thetransconformation of a doubly substituted ethylene possesses a lower total energy and thus is more stable than itsciscounterpart.The reason behind lies in the fact that thetransconformer is energetically more favorable in steric and electrostatic interactions.There are exceptions,however.Halide-containing species,such as 1,2-difluoroethylene,1,2-dichloroethylene,and 1,2-chlorofluoroethylene,are among these exceptions.Their unusual stability in thecisconformation ranging from 1.09 to 4.52 kJ·mol-1as determined by experiments1-5is likely due to the strong electronegativity of halide atoms.This irregular stability in thecisisomer is often called thecis-effect.A double bond is not a prerequisite for this effect since a similar“gauche effect”is found in 1,2-difluoroethane6,7and 1,2-dimethoxyethane8between the gauche and anti-rotamers.While it was Craig1who first experimentally demonstrated the existence of thecis-effect,many studies have been carried out to ascertain the existence of this effect.Nonetheless,no consensus about its origin and nature has been available in the literature.
Using molecular orbital theory,Epiotis9established an approach to categorize the non-bonding interactions,proposing that the interaction between the two lone pairs of the fluorine atoms in 1,2-difluoroethylene is attractive,thus rendering more favorable interactions in thecisisomer.Later,Kollman10carried outab initiocalculations,based on which he argued that the attraction in 1,2-difluoroethylene is not from the lone pair interaction.Rather,it came from the changing characteristic of the C-F single bond due to the more electronegative nature of fluorine.Bemardiet al.11elucidated that the overall impact of the interactions in orbitals or lone pairs is destabilizing.Instead,it is the total less destabilizing interaction in thecisisomer than in thetransisomer that makes the effect valid.
Meanwhile,Cremer12has showed that with the MP2 method,the standard Pople's polarized 6-31G(d),6-31G(d,p),and 6-311G(d)basis sets,and the experimentally determined geometries,13a satisfactory energy difference of 3.77 kJ·mol-1between thetransandcisconformers of 1,2-difluoroethylene can be obtained,suggesting that the electron correlation can be used to account for the small energy difference between the two conformations.Gandhi and co-workers14performed a series of calculations with correlated basis sets from DZ to TZ+P augmented with diffusion functions for the system and the results indicated that as long as an adequately large basis set and accurate experimental geometry are adopted,thecis-effect can be correctly reproduced even at the Hartree-Fock level of theory.Notice,though,that the information of correlation effects is implicitly contained in the experimental geometry.In addition,Saebøet al.15optimized thecisandtransforms of 1,2-difluoroethylene at the level of Møller-Plesset fourth-order perturbation theory by using the 6-311G basis augmented with two sets of polarization functions.Their results reveal that to properly describe thetrans-cisenergy difference,both accurate molecular geometries and large atomic basis sets with diffuse functions included are necessary and the difference in the correlation energy between thetransandcisisomers is sensitive to the geometries employed but negligible when evaluated at the relaxed geometry.Dixonet al.16pointed out that the primary problem of Cremer12and Gandhi14was the strong dependence of their results on the experimental geometry.There are three sets of experimental structural data in the literature and there is no consensus on which one is to use.If the geometry is from the optimized structure of the same method,Dixonet al.16discovered that the electron correlation played a negligible role for thetrans-cisenergy difference of 1,2-difluoroethylene.The most recent work about thecis-effect by Yamamotoet al.17has employed high levelab intioand density functional theory(DFT)approaches and indicated that the lone pair electron delocalization has played the role of reducing intramolecular interactions.
In this work,we revisit this unresolved problem and tackle it with a number of new approaches that have become available only relatively recently.Our objectives are two-fold.At first,we unambiguously demonstrate the existence of thecis-effect with a list of density functionals at different levels of theory and a selection of basis sets.Secondly,employing a number of analysis tools such as the natural bond orbital(NBO)analysis,energy decomposition analysis(EDA),density functional reactivity theory(DFRT),and non-covalent interaction(NCI)analysis,we investigate the nature and origin of thecis-effect.We find that there exists a weak but attractive non-covalent interaction between the two substituting atoms/groups in thecisconformer.We also find that the electrostatic,steric,and kinetic energy contributions all play important roles for the validity of thecis-effect,but none of these quantities can be used alone to explain the general validity of the effect,suggesting that thecis-effect is a complicated one,resulted from compound interactions from a number of interactions.Results from NBO,EDA,DFRT,and NCI provide a consistent picture about the validity and origin of thecis-effect from the present study.Instead,we employed two-variable explanations to justify the general validity of this effect using the electrostatic interaction as the primary term plus either the steric effect or kinetic energy component.
2 Methodology
In DFT,the total electronic energy of a system,E[ρ],can be expressed in terms ofTs[ρ],Ee[ρ],andExc[ρ],18

whereTs[ρ]stands for the non-interacting kinetic energy,Ee[ρ]denotes the electrostatic energy,andExc[ρ]represents the exchange-correlation energy component andρis the electron density distribution.The electrostatic term consists of the nuclear to electron attraction,Vne[ρ],the classical inter-electron Coulombic repulsion,J[ρ],and the nuclear-nuclear repulsion,Vnn.

Recently,one of the present authors proposed an alternative energy decomposition scheme to quantify the steric hindrance within the DFT framework by using the Weizäcker kinetic energy,Tw[ρ]19
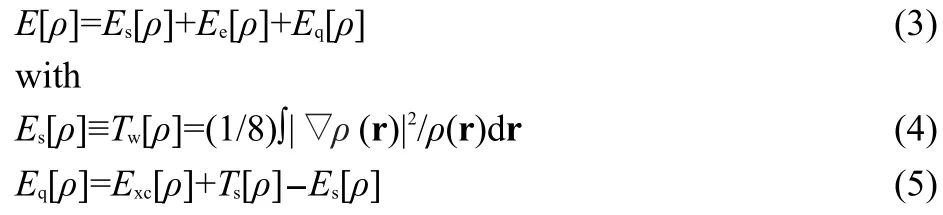
whereEsandEqare the energy contributions from the steric and fermionic quantum effects,respectively,ρ(r)is the total electron density,and ▽ρ(r)is the electron density gradient.This novel quantification of the steric effect has been applied to a number of systems,such as conformational changes of small molecules,20SN2 reactions,21chained and branched alkanes,22experimental electron densities of crystals,23and the anomeric effect.24Reasonably good trends and linear relationships between theoretical and experimental scales of the steric effect have recently been observed25by Taft at both group and entire molecular levels.In the current contribution,this new energy decomposition scheme together with the conventional approach will be utilized to analyze the origin of thecis-effect,by obtaining the total isomerization reaction energy and its components to find out which component(s)is/are the dominant term(s)contributing to the validity of thecis-effect.
The non-covalent interaction(NCI)analysis,proposed recently by Yang and co-workers,26-28is stemmed from the electron density and its first-order derivative,s=1/(2(3π2)1/3)|▽ρ(r)|/ρ(r)4/3,which is a dimensionless quantity in density functional theory to depict the deviation from a homogeneous electron density distribution.The reduced density gradient(RDG),defined bys,possesses very large positive values in the regions far from the molecule,but it becomes small values in the vicinity of both covalent and non-covalent interactions.This quantity has been employed to identify weak non-covalent intra-and inter-molecular interactions.26,29To identify different types of interactions,the sign of Laplacian of the density, ▽2ρ,is often used,30which is usually reduced into the sum of contributions along the three principal axes of its maximal variations.These components are the three eigenvaluesλiof the electron-density Hessian(second-derivative)matrix,such that ▽2ρ=λ1+λ2+λ3(λ1≤λ2≤λ3).We chose the quantityλ2over the Laplacian in this work.Plotting low-gradient isosurfaces,subject to a further low-density constraint,enables real-space visualization of noncovalent interactions,even at the DFT B3LYP level of theory.31We make use of this quantity in this work to detect weak interactions in thecisconformation.In addition,to understand the origin of the effect,we will also employ reactivity descriptors from density functional reactivity theory(DFRT),18,32,33also called conceptual DFT,to identify possible correlations between energy differences of thetransandcisisomers and these indices,including chemical potential,hardness,condensed Fukui functions,electrophilicity,fugality,etc.18,32,33
3 Computational details
We choose twelve doubly substituted ethylene compounds in this study(Scheme 1),such as 1,2-difuoroethylene,1,2-dichloroethylene,1,2-dimethoxyethylene,1,2-chlorofluoro ethylene,and 1,2-chloromethylethylene,whose experimental data have unambiguously validated that these systems prefer to be in thecisconformer in the ground state.1-5The last three systems in Scheme 1 are normal species whosetransisomers are lower in energy than theirciscounterparts.They are included here for the comparison purpose.Bothtransandcisisomers of these species will be considered in this study.Different density functional and basis sets will be used to optimize both thecisandtransforms of these systems to verify the validity of thecis-effect.The standard Pople's basis set of split-valence triplezeta plus(d,p)-like polarization and diffusion functions,6-311+G(d,p),in conjunction with a set of exchange functionals:S,XA,B,PW91,mPW,G96,PBE,O,TPSS,BRx,PKZB,wPBEh,PBEh,and a sequence of correlation functionals:VWN5,LYP,PL,P86,PW91,B95,TPSS,KCIS,PKZB,and VP86(VWN5 local and P86 non-local correlation functional),will be used for the purpose.Following exchange-correlation density functional forms,HFS,XAlpha,HFB,HCTH407,M06L,B3LYP,M05,M052X,M06,M06HF,M062X,O3LYP,and X3LYP will be employed to evaluate the total energy differences with the following standard Pople's basis sets,6-311G(d,p),6-311+G(d,p),and 6-311+G(2d,2p),as well as Dunning's augmented correlation consistent basis sets,aug-cc-pVDZ and aug-ccpVQZ.34The effective core potential(ECP)basis set,LANL2DZ,35was also tested.
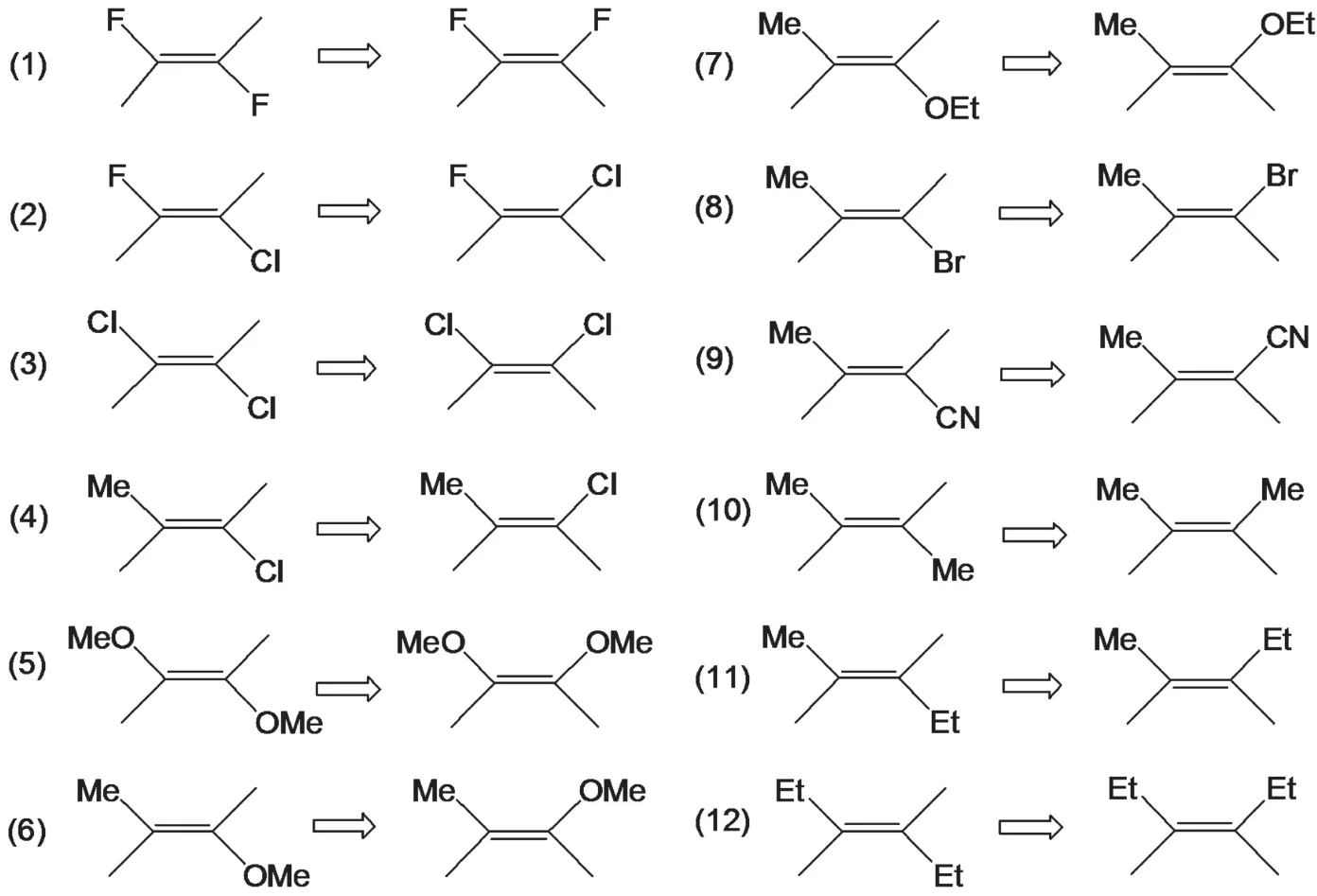
Scheme 1 trans and cis conformations of the twelve doubly substituted ethylenes studied in this work
All quantum mechanical calculations have been carried out using Gaussian-09 version B.01 package36with no symmetry constraint,tight self-consistent field(SCF)convergence criteria,and ultrafine integration grids.After a full geometry optimization,a single point frequency calculation was performed to verify that the structures obtained were indeed a minimum on the potential energy surface(e.g.,no imaginary frequency).A full natural bond orbital(NBO)analysis37for 1-center atomic hybrids was subsequently performed at the DFT B3LYP/augcc-pVDZ level of theory to acquire the second-order perturbation energy from the Fock matrix.As thetrans/cisenergy difference is small,the default printed energy threshold 2.09 kJ·mol-1was changed to 0.04 kJ·mol-1through the keyword E2PERT.NWChem6.038was employed to perform the energy decomposition analysis,which was conducted at the DFT B3LYP/aug-cc-pVDZ39level of theory.As a comparison,we also calculated the steric energy from the wave function theory using natural orbital analysis for all the systems.The NBOFILE keyword in NWChem was utilized to create an output file to be used as the input file for the stand-alone natural bond orbital analysis code,NBO version 5.0.40We denote the result from this calculation as the NBO steric energy41and the difference denoted by ΔNBOSteric.NCIPLOT2.042was used to generate grid points for non-bonded weak interactions within the molecules.VMD1.943was used for the visualizations of the reduced density gradient(RDG)versussign(λ2)ρ(r).The AIMAll software44was used to anaylze the wavefunctions generated at the DFT M062X/aug-cc-pVDZ level of theory.The envelope size of 0.001 electrons·Bohr-3isodensity was employed.
4 Results and discussion
Table 1 lists the calculated and experimental energy differences(ΔE=Etrans-Ecis)between thetransandcisisomers for the 12 species studied in this work.The calculated values were obtained at six levels of theory,DFT(B3LYP),DFT(M062X),MP2,MP4,QCISD(T),and CCSD(T)with the aug-cc-pVDZ basis set and the geometry obtained at the DFT B3LYP/6-311+G(d,p)level of theory.From Table 1,one can see for these nine compounds with thecis-effect in effect that(i)both experimental data and computational results have firmly established theexistence of thecis-effect,that is,thecisisomer is more stable(ΔE>0)than itstranscounterpart;(ii)qualitatively speaking,all theories are able to reproduce the experimental results,giving in all the cases that ΔE>0 for these 9 compounds;(iii)in general MP2 and CCSD(T)results are closer to the experimental data than DFT results but there are exceptions such as compounds 3 and 5 with reasons unknown;(iv)QCISD(T)is slightly better than CCSD(T)in accuracy when compared with experimental data,whose reason behind is also unknown;and(v)B3LYP and M062X functionals give similar results in quality.Because of these reasons,we employ both B3LYP and M062X functionals in our later analyses.For the systems where nociseffect comes into play,we find that all DFT andab initioapproaches were both able to predict that thetransisomer is more stable.
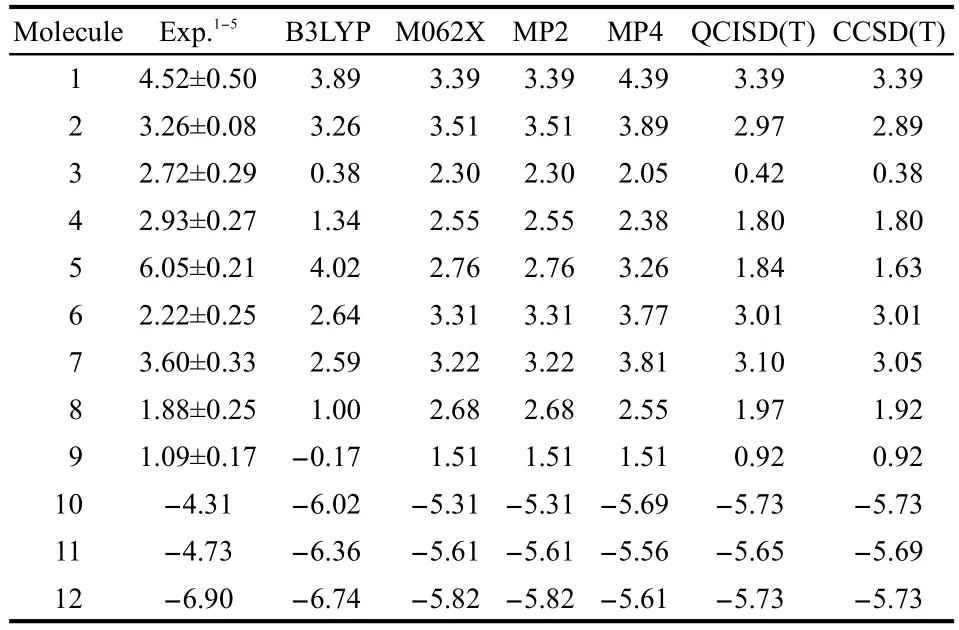
Table 1 Experimental energy differences(Etrans-Ecis)(unit in kJ·mol-1)between the trans and cis isomers,and calculated energy differencesat different levelsof theory[DFT,MP2,MP4,QCISD(T),and CCSD(T)]for the 12 systems studied in this work
As an example,Table 2 shows the structural,electronic,and spectroscopic properties of both thecisandtransconformers of the 1,2-difluoroethylene from the B3LYP/aug-cc-pVDZ level of theory with the structure obtained at the B3LYP/6-311+G(d,p)level of theory as well as some geometrical parametersfrom Felleret al.45.We find that the C-F and C-H bond lengths in thecisisomer are shorter than those in thetranscase but the C=C bond becomes longer.This elongated C=C bond in thecisisomer allows the∠F-C=C angle to be increased by 2.6°without affecting the∠F-C-H angle.Thermochemistry data of these two species in Table 2 indicate that thecisisomer is 3.30 kJ·mol-1lower in energy and 5.19 kJ·mol-1lower in Gibbs free energy than itstranscounterpart.This extra stability of thecisisomer confirms the existence of thecis-effect for this molecule.On the other hand,this result suggests that entropy also contributes positively to the stability of thecisconformer.From the IR/Raman spectral results,we find that though the anti-symmetric C-F stretching in thecisisomer is weaker than that in thetransisomer,stronger peaks in the in-plane and out-of-plane bending modes of the∠F-CH angle are observed in thecisform.These stronger bending modes come from the extra stability of thecisform.We also see a blue-shifted peak for the symmetric C-H stretching in thecisform.TD-DFT calculations show that the first excitation peak is red-shifted in thecisform,compared to that of thetransform,indicating that even though thecisisomer is more stable,it possesses a relative smaller HOMO/LUMO(the highest occupied molecular orbital/the lowest unoccupied molecular orbital)gap.This is confirmed by the HOMO/LUMO results in Table 2,where one can find that the HOMO/LUMO gap in thecisform is narrowed by 0.015 eV relative to thetransform.Looking at the HOMO and LUMO energies,we find that they are both lowered in thecisform,just that HOMO is lowered relatively smaller than LUMO,making the HOMO/LUMO gap is a little smaller than that of thetranscounterpart.Taking a look of the NBO charge on the atoms,we find that,relative to thetransform,C is less positively charged and F is less negatively charged,suggesting that there exist more covalent contributions in the C-F bonds in thecisisomer.Meanwhile,H is more positively charged,indicating that the stronger C-H bond in thecisform possesses more ionic feature,as indicated by the shorter C-H bond and enhanced C-H symmetric stretching in Table 2.

Table 2 Structural,electronic,and spectroscopic properties of the two isomers of 1,2-difluoroethylene
Next,we examine the impact of exchange-correlation energy density functionals and basis sets on the validity of theciseffect.Again,we use 1,2-difluoroethylene as an example.Table 3 shows thetrans-cisenergy difference for a series of local and nonlocal exchange functionals,S,XA,B,PW91,mPW,G96,PBE,O,TPSS,BRx,PKZB,wPBEh,and PBEh combined with different categories of correlation functionals,VWN5,LYP,PL,P86,PW91,B95,TPSS,KCIS,PKZB,and VP86.We employed the standard Pople's basis set 6-311+G(d,p)for these tests.From Table 3,one can find that although there exist quantitative differences in numbers,in all the cases the energy difference is positive,indicating that thecis-effect is always in place and its validity does not depend on the choice of approximate exchange-correlation energy density functionals.With the same basis set,theab initioCCSD(T)method gives thetrans-cisenergy difference of 3.39 kJ·mol-1.
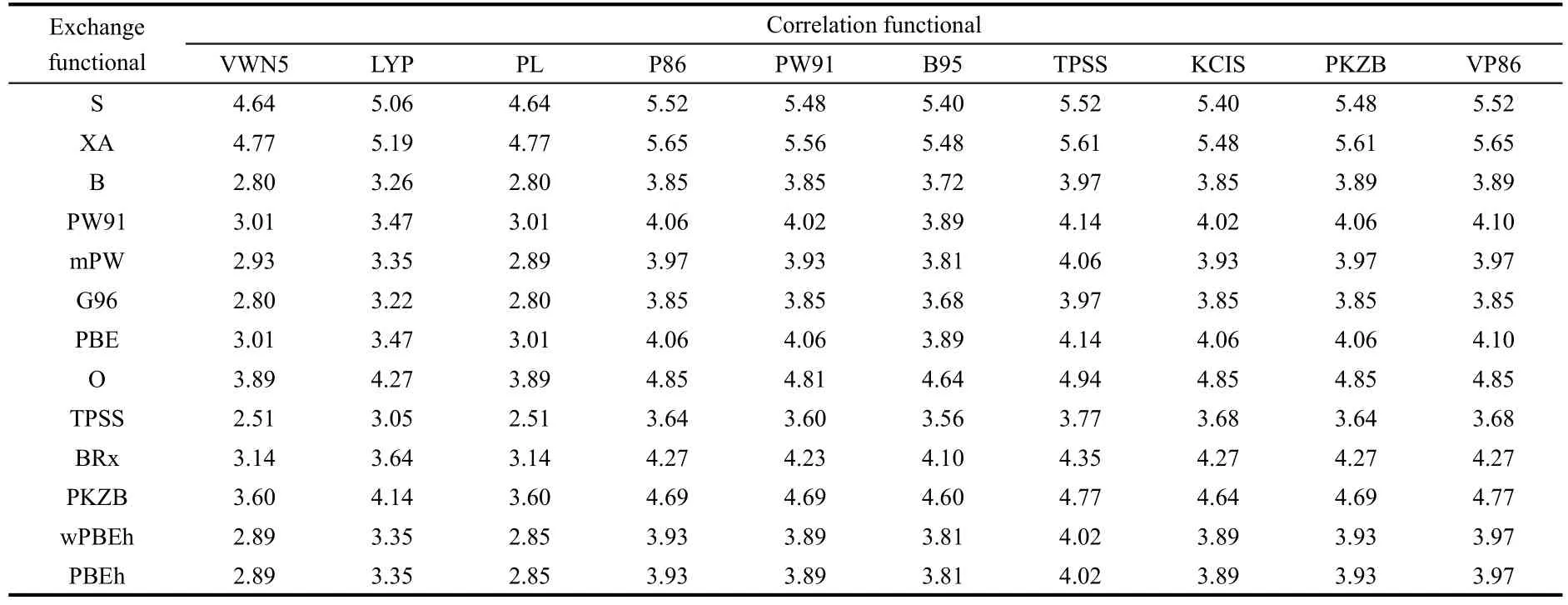
Table 3 Total energy difference(unit in kJ·mol-1)between the trans and cis isomers for 1,2-difluoroethylene from combinations of different exchange and correlation functionals and the standard Pople's basis set,6-311+G(d,p)
Table 4 exhibits the dependence of thetrans-cisenergy on the basis set for a list of approximate functionals.Effective core potential basis sets such as LANL2DZ fail to reproduce thecis-effect in almost all the tests,whereas high-level Dunning's augmented correlation consistent basis sets such as aug-ccpVDZ and aug-cc-pVQZ are able to predict correctly and quantitatively.The failure of the LANL2DZ ECP basis set reminds us of the paramount importance of the impact from inner electrons.Using standard Pople's basis sets,we obtained mixed results.It appears that with the addition of polarization and diffuse functions in basis sets the prediction results become more reliable.Put together,these results in Tables 3 and 4 demonstrate that the validity of thecis-effect is less likely to depend on the choice of approximate functionals but more on the choice of the basis set.A high-level basis set is recommended when this effect is studied.We employ the B3LYP functional and aug-cc-pVDZ basis set for the rest study in this work.
Now let us find out where the extra stability of thecisisomer comes from.To that end,we first make use of the following two analysis tools.The first tool is the conventional NBO analysis and the other is the relatively new approach through the non-covalent interaction(NCI)analysis to identify noncovalent interactions within a molecule.The NBO analysis was done by performing the second-order perturbation theory analysis of the Fock matrix in the NBO basis to estimate the stabilization energy(E2)associated with the delocalization from a donor molecular orbitalito an acceptor molecular orbitaljasE2≡ΔEij=qi×F(i,j)2/(εj-εi),whereqiis the donor orbital occupancy,εjandεiare the orbital energies,andF(i,j)is the off-diagonal Fock matrix element expressed in the NBO basis.This quantity is a quantitative indication of nonbonding interactions and thus the extra stability gain between occupied and unoccupied molecular orbitals,which is also called the hyperconjugation effect in organic chemistry.Table 5 shows the total second-order perturbation energy values,E2,computed with B3LYP and M062X functionals between the two substituting atoms or groups in bothtransandcisconformations of the 12systems.Using the NOBOND keyword,we only considered 1-center atomic hybrids in this study.As can be seen from Table 5,the following three points are in order:(i)there exist hyperconjugation interactions between the two substituting atoms/groups in both thecisandtransconformations;(ii)this kind of interaction is stronger in thecisisomer than in thetransform;and(iii)the difference of these energies,ΔE2,between the two conformations sometimes is less than the total energy difference ΔE,ΔE2<ΔE,for some systems such as FHC=CHF and FHC=CHCl,whereas in other cases such as MeHC=CHCl and MeOHC=CHOMe,we have ΔE2>ΔE.These NBO analysis results suggest that there is a stronger noncovalent interaction between the two substituting atoms or groups in thecisconformer and this extra stability in some systems can be used to explain its extra stability and thus the validity of thecis-effect for them.However,in other systems,the magnitude of this extra stability is not big enough to account for the validity of theciseffect.More importantly,the last 3 examples in Table 5,where thecis-effect does not come into play,also have a largerE2(cis)thanE2(trans),suggesting that using the hyperconjugation interaction alone,one cannot explain the general validity of thecis-effect.
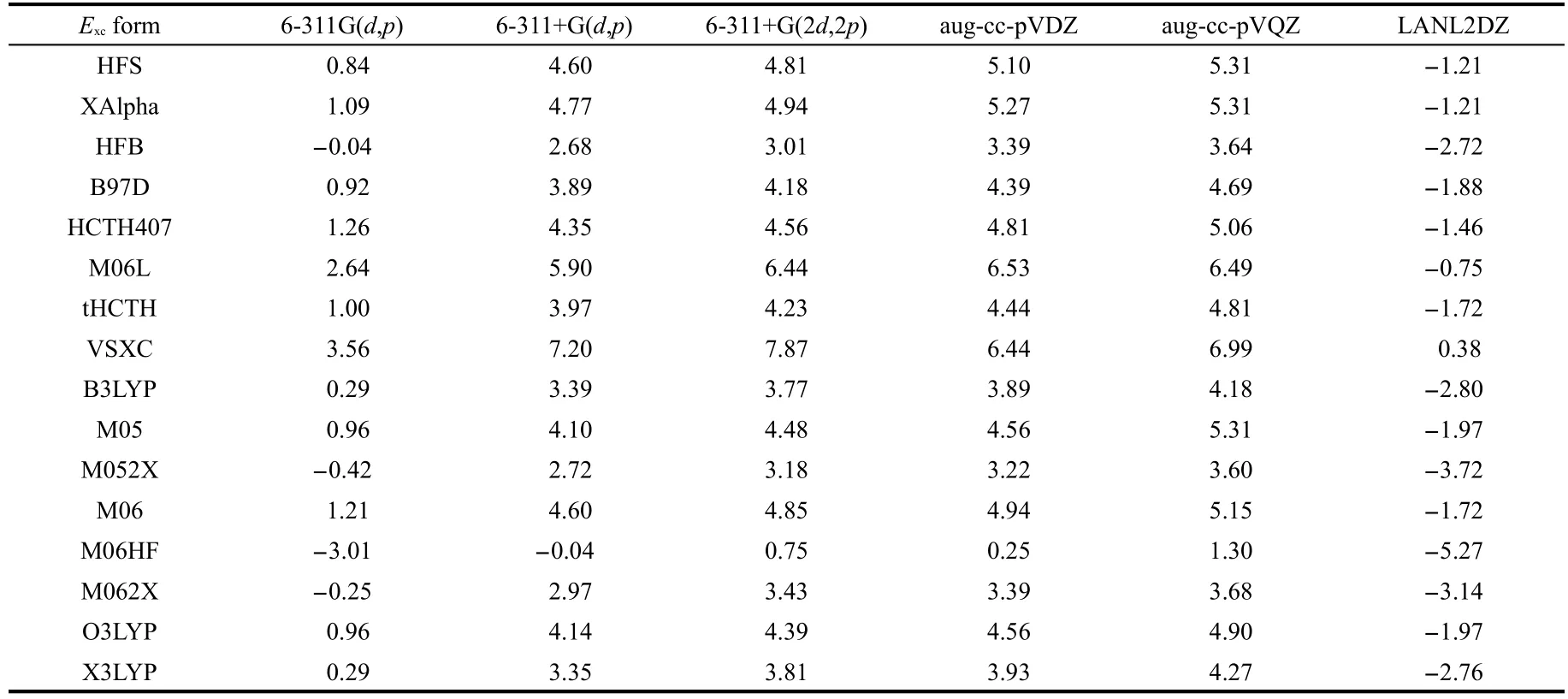
Table 4 Impact of basis sets on the total energy difference(unit in kJ·mol-1)between the trans and cis isomers for 1,2-difluoroethylene
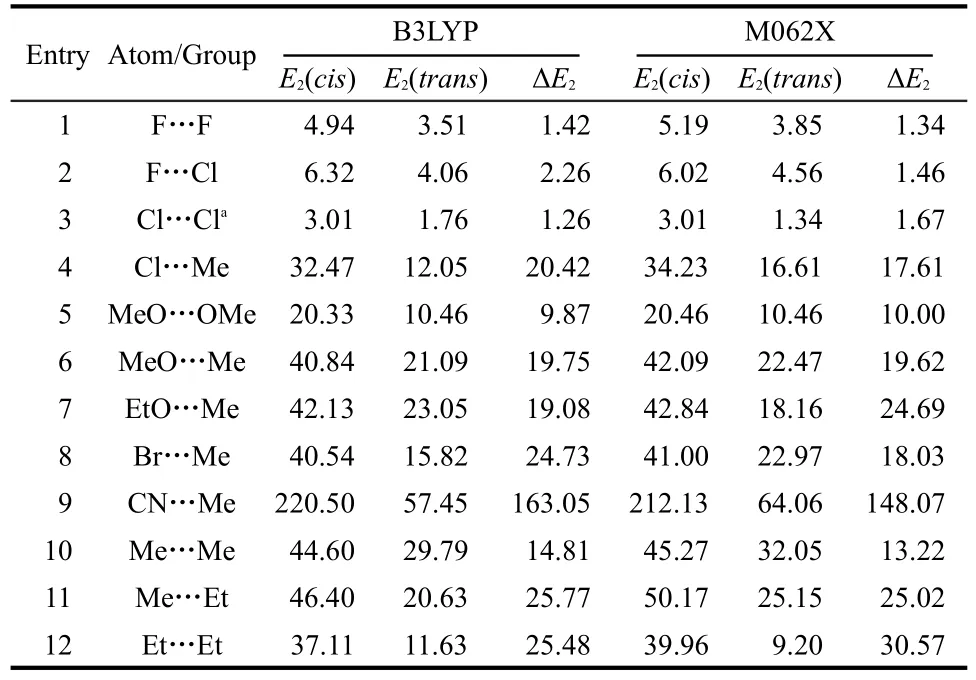
Table 5 Acomparison of the second-order perturbation stabilization energy(unit in kJ·mol-1)from the one-center atomic hybrids between the two substituting atoms or groups for both trans and cis conformations of the 12 species studied in this work
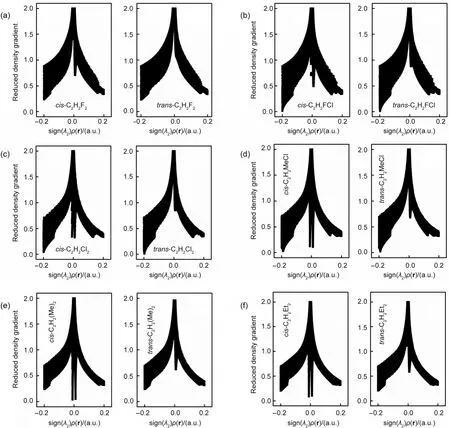
Fig.1 Diagrams of the reduced density gradient(RDG)versus sign(λ2)ρ(r)for both the cis and trans conformations of a few molecules with the cis-isomer shown on the left and the trans-isomer on the right
The fact that there exist nonbonding interactions between substituting groups in thecisisomer can be further witnessed from the NCI analysis.Shown in Fig.1 as illustrations are the reduced density gradient(RDG)diagrams for bothcisandtransisomers for six of the systems studied in this work,the first 4 systems(Fig.1(a-d))with thecis-effect in action and the remaining 2 systems(Fig.1(e,f))as the comparison without the effect in place.Fig.1a shows the RDGvssign(λ2)ρ(r)for bothcisandtransforms of C2H2F2from the B3LYP/6-311+G(d,p)density.As can be seen,there exists a conspicuous“spike”in the proximity of zero density for thecisconformation,unambiguously confirming the existence of weak,non-covalent interactions in thecisform.Such a“spike”,however,never showed up in thetransisomer of C2H2F2.The same observation of the density spike in thecisisomer can be found for other compounds,Fig.1(b-d),indicating that there exist stronger weak non-covalent interactions between substituting atoms or groups in thecisconformation,which might lead to the extra stability of the isomer.For their correspondingtransisomer,even if such spikes exist,they are much smaller in size.Nevertheless,in Figs.1(e)and 1(f)for C2H2Me2and C2H2Et2,respectively,in which nocis-effect is present,the same pattern of the“spikes”is observed.These results suggest that even though we found stronger week interactions between substituting groups in thecisisomer they cannot be employed to justify the validity thecis-effect,agreeing with what we observed in the NBO analysis.
Another way to present the RDG result is to employ the isosurface.Fig.2 shows such isosurfaces for the same set of molecules plotted at the isovalue of 0.75 a.u.at the same level of theory.In each case,a small disc-like domain between the two substituting atoms or groups is clearly seen,with red areas in the disc representing repulsive interactions and blue regions representing attractive interactions.These isosurfaces confirm once again that there exist weak interactions between these two structural motifs and thecisisomer possesses stronger interactions.These nonbonding interactions,however,could be both attractive and repulsive,verifying why they cannot be employed to explain the general validity of thecis-effect.
Hereafter,we switch the gear to the energy decomposition analysis.In Table 6,we perform energy decomposition analysis using Eqs.(1)and(3),respectively.According to these formulas,the total energy difference ΔEbetween thetransandcisisomers can be decomposed into three independent contributions as
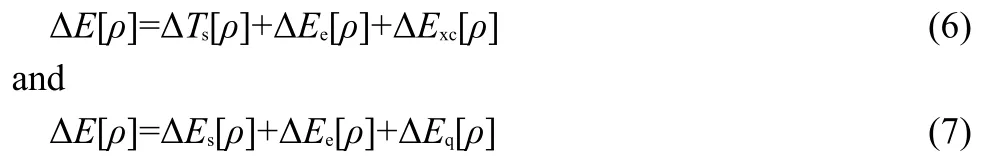
In the first decomposition scheme,ΔEcomes from three contributions:kinetic ΔTs,electrostatic ΔEe,and exchange-correlation ΔExc,whereas in the second scheme,the three components are steric ΔEs,electrostatic ΔEe,and fermionic quantum ΔEq.The common term in both schemes is the electrostatic contribution ΔEe.Columns 3 to 7 of Table 6 display the results of these two analyses using the M062X approximate functional and the aug-cc-pVDZ basis set.From Table 6,we can see that among the five energy components,ΔEqis always negative,ΔEq<0,ΔExcis always positive,ΔExc>0,ΔEs>0 except for the first molecule,and ΔTsand ΔEecan be either negative or positive.To explain why ΔEis positive in Table 6,ΔE>0,for the first 9 systems and negative,ΔE<0,for the rest 3 species,if there is only one single energy component to dictate the validity of theciseffect in all the cases,one would require to have a quantity in Table 6 whose sign either goes together with or is inverse of ΔE.Unfortunately,as is apparently shown from Table 6,there is no such a quantity.The closest two quantities that one can spot from Table 6 are ΔTsand ΔEe,where ΔEehas the same sign as ΔEin general and ΔTsoften possesses the opposite sign as ΔE.Fig.3(a)plots the correlation between ΔEevsΔE,where one finds that the correlation coefficientR2is 0.71.WithTs,we obtainR2=0.67(plot not shown).Even though these correlations are reasonably good,as seen from Table 6,both quantities have exceptions.For example,for ΔEe,compounds 3,4,8,and 9 are exceptions,and for ΔTs,compounds 4,8,and 9 are outliers.In this regard,Table 6 shows that there is no single energy component solely governing the general validity of thecis-effect.
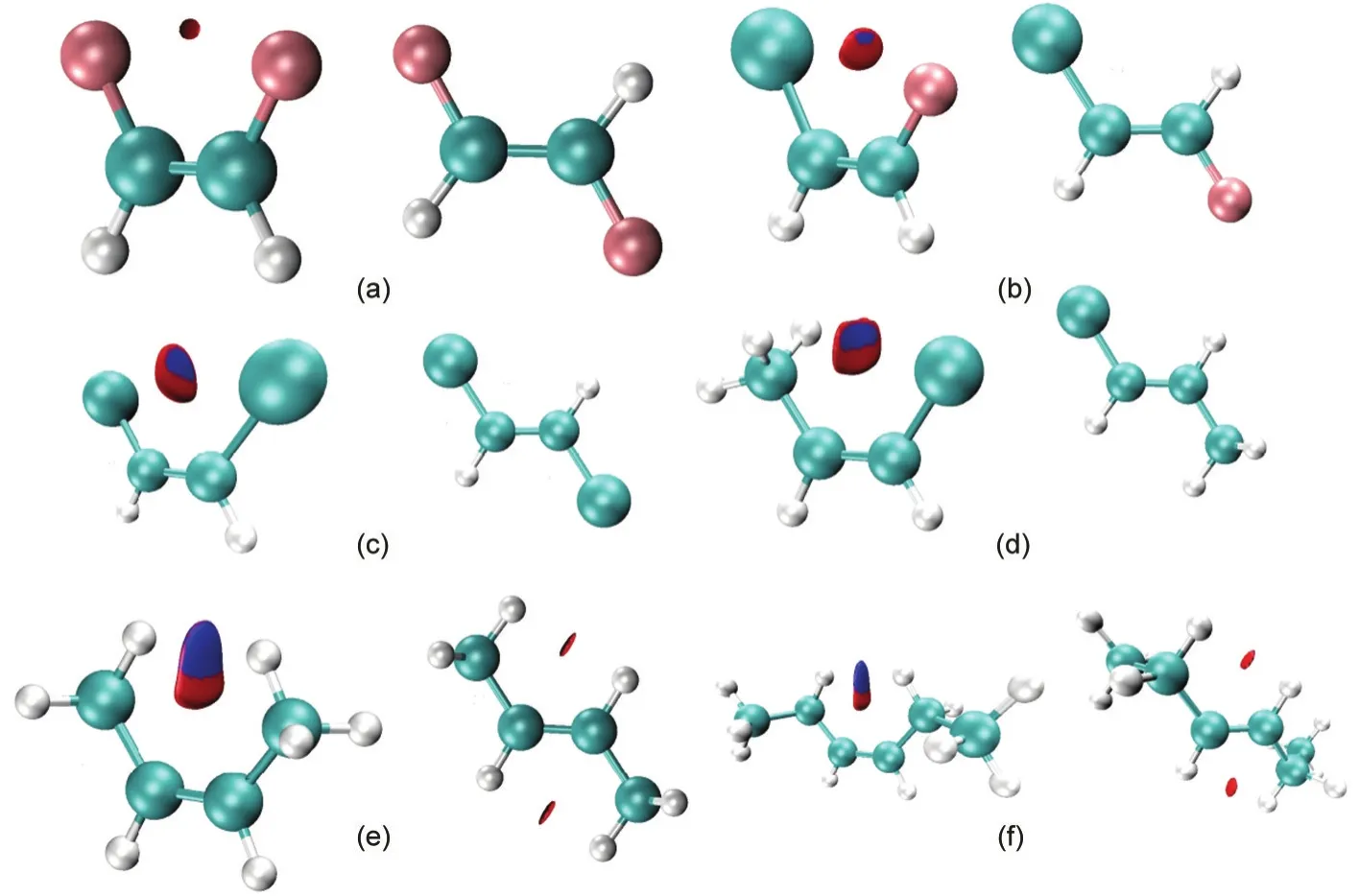
Fig.2 Reduced density gradient isosurfaces for both the cis and trans conformations for six of the systems studied in this work
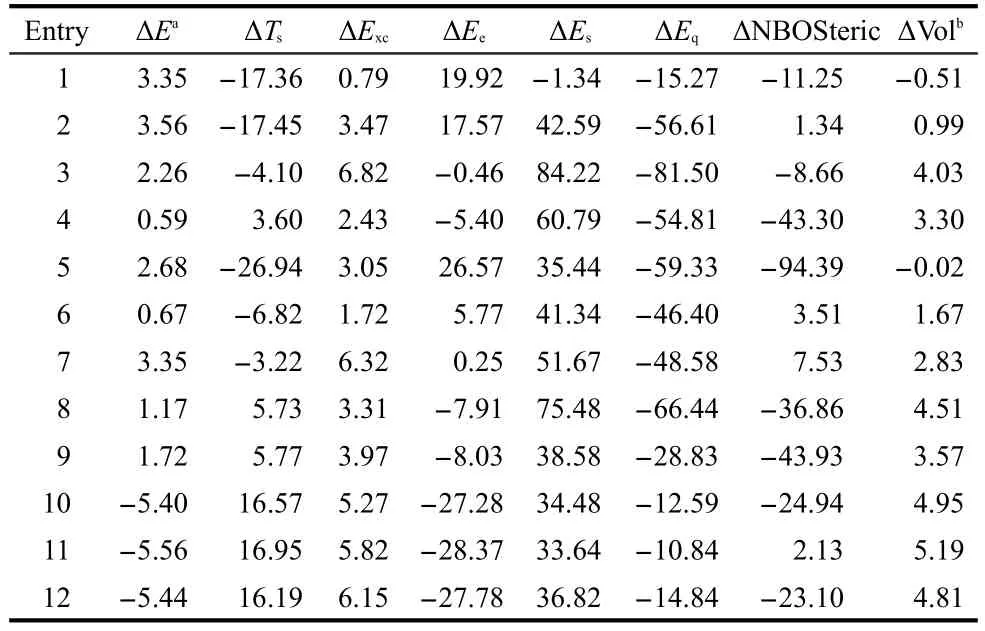
Table 6 Results of two energy decomposition analysis schemes with the ΔNBOSteric,kinetic(ΔTs),steric(ΔEs),exchangecorrelation(ΔExc),electrostatic(ΔEe),fermionic quantum(ΔEq),and total(ΔE)energies for the trans/cis isomerization reactions in Scheme 1 with the M062X functional and aug-cc-pVDZ basis set(unit in kJ·mol-1except the molar volume difference,ΔVol,cm3·mol-1)
That is to say,the existence and validity of thecis-effect is a complicated phenomenon,suggesting that more than one interaction are involved.In Figs.3(b)and 3(c),we employ two-variable explanations from Eqs.(6)and(7).The least-square fitting using the two quantities for each of the two energy decomposition schemes gives better fits to the calculated total energy difference,with the correlation coefficientR2equal to 0.86 and 0.87,respectively.As can be seen from Table 6 and the fitted formulas in the Figure,ΔEeis a key component.These results demonstrate that,even though there are exceptions,ΔEeis the most important contributor to the validity of the effect.A positive ΔEefor the cases where the effect comes into play suggests that thecisisomer has larger electrostatic interactions(Eeitself is a negative quantity),whereas in normal systems,it is thetransisomer that possesses the larger electrostatic interactions.In other words,if a ΔEeexplanation is true,the lower the electrostatic energy the lower the total energy in both normal and abnormal cases.In Fig.3(b),ΔTsis the second factor contributing to the fitting,whereas in Fig.3(c),ΔEsis the other component in the least-square fit.With these fits,we found no exception from Fig.3(b),and only one except was observed in Fig.3(c),indicating that much better results can be obtained with the scenario of using two parameters to explain the general validity of thecis-effect.We tried other combinations of two-variable fits and no better fitting result was obtained.
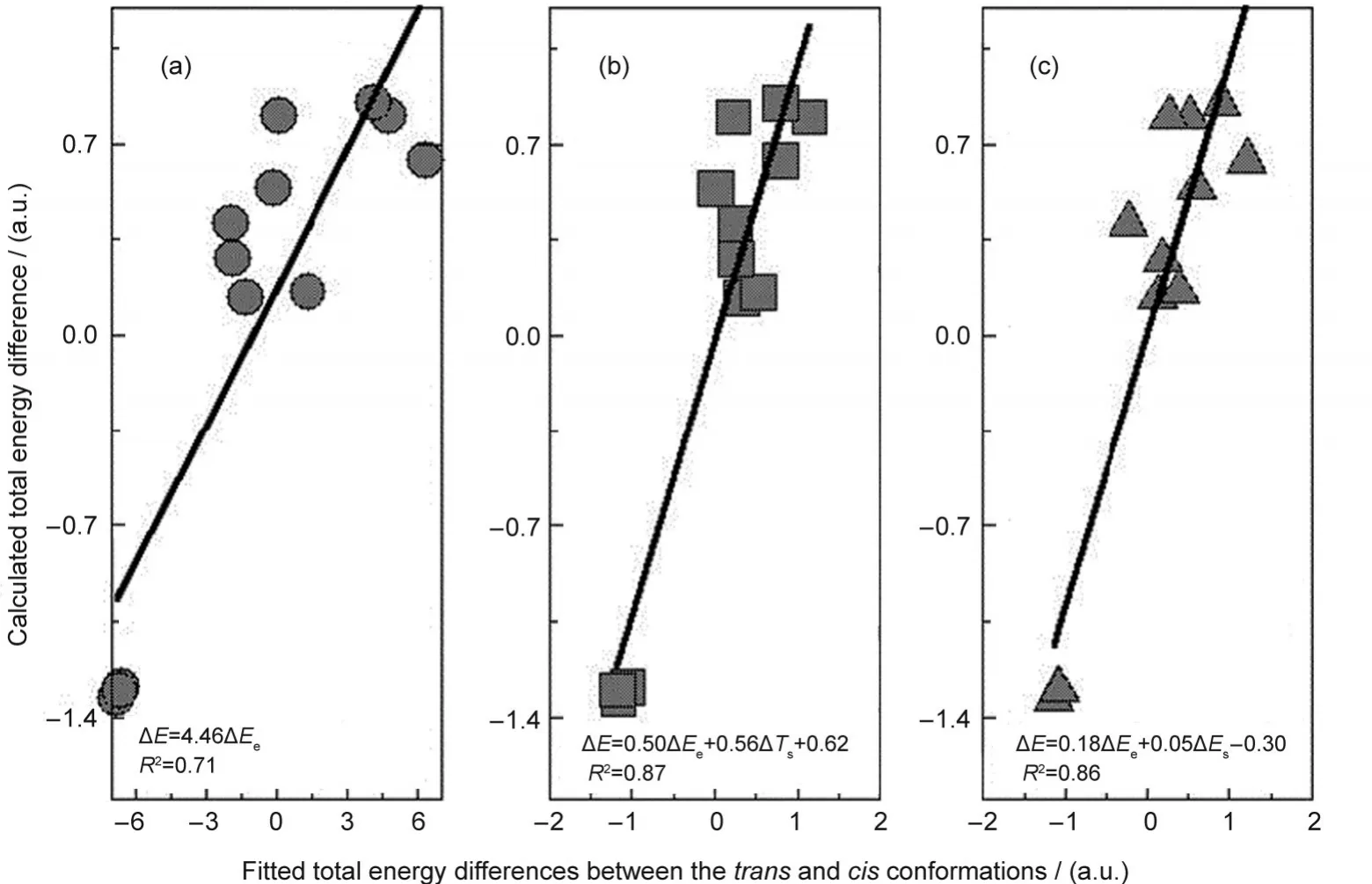
Fig.3 One-and two-variable least-square fittings for the total energy difference using different energy components
In Table 6,some other interesting features can be seen.The first one is ΔExc,which is always positive,even though ΔEitself can be both positive and negative.A positive ΔExcshows that thecisisomer possesses a larger exchange-correlation in-teraction(Excis a negative quantity).The second one is the always negative ΔEq,the fermionic quantum energy component,which itself is a positive quantity.An always negative ΔEqsuggests that the fermionic quantum repulsion is larger in thecisisomer than in thetransform.Finally,it is the DFT steric energy difference,ΔEs,which is positive except for the first molecule.A positive value of ΔEsmeans a smaller steric repulsion in thecisisomer becauseEsis always non-negative.SinceEsis homogeneous of degree one in density scaling,46this quantity is positively proportional to the molecular size(or volume).To verify this statement,listed in the last column of Table 6 is the molar volume difference of thetransandcisisomers of these systems.From Table 6,we find that ΔVol is positive in 10 out of 12 cases and it has the same sign as ΔEsexcept for compound 5,which has a very small value of ΔVol.These results demonstrate that thecisisomer always has a smaller size than thetranscounterpart in most of the cases and are in good agreement with our ΔEsresults.
For the purpose of comparison,also shown in Table 6 is the result of the wavefunction-theory-based quantification of the steric effect from the NBO analysis,ΔNBOSteric,which uses the Pauli Exclusion Principle to account for repulsions among same spin electrons.As can be seen from Table 6,ΔNBOSteric can be either positive or negative with no correlation to ΔEat all,suggesting that this quantity cannot be used to explain the validity of the effect.We have also considered other possible quantities for these systems with structural and electronic properties from DFT or density functional reactivity theory(DFRT)frameworks,such as changes in bond lengths,NBO charges,molecular electrostatic potential,47natural atomic bond energies,chemical potential,hardness,electrophilicity,etc.18,32,33No consistent pattern from these examined quantities has been identified for these species(results not shown).
Put together,our present results from the various computational and analysis tools suggest that(i)DFT is able to computationally verify the validity of thecis-effect,it is less dependent on the choice of approximate functionals and more on basis sets;(ii)NBO and NCI analyses unambiguously show that there exists a stronger nonbonding interaction between the two substituting atoms or groups in thecisisomer,but this kind of interactions could be both attractive and repulsive and is not big enough in many cases to account for the validity of theciseffect;(iii)energy decomposition schemes present a complicated picture about the behaviors of different energy components,where no single interaction was found to be solely responsible for the validity of thecis-effect,with ΔEegiving the best fit ofR2=0.71;(iv) ΔExc>0,ΔEs>0,and ΔEq<0,meaning that thecisisomer always has larger exchange-correlation interaction,less steric repulsion and stronger fermionic quantum repulsion,with the less steric repulsion verified by the less molar volume;and(v)with two-variable fits,good correlations withR2around 0.85 were obtained with either ΔEeand ΔTsor ΔEeand ΔEs.These results confirm that thecis-effect is a complicated phenomenon with multiple origins and one cannot simply explain its general validity with one single interaction.To the best knowledge of the present authors,it is the first time that this complicated origin and nature of thecis-effect for these doubly substituted ethylenes is carefully analyzed and revealed.We expect that the findings obtained from this work are readily applicable to other similar systems as well.
5 Concluding remarks
In summary,in this work,the nature and origin of theciseffect dictating the stability of a number of doubly substituted ethylenes are investigated,where we find that it is rather a complicated phenomenon and cannot simply employ one single interaction to explain its validity.Conventionally,thetransisomer of a doubly substituted ethylene should be more stable than itsciscounterpart because of the more favorable electrostatic and steric interactions in thetransconformer.However,there are many exceptions such as 1,2-difluoroethylene and 1,2-dichlorethylene.The abnormal stability of thecisisomer of these doubly substituted ethylene compounds is called theciseffect.To understand its nature and origin,which are still not clear,in this work,using twelve simple molecules as examples,XHC=CHY(X,Y=F,Cl,Br,CN,CH3,OCH3,C2H6),9 of which exhibit thecis-effect with the remaining 3 systems as conventional systems used for the comparison purpose,we have systematically investigated the validity,nature,and origin of this effect.A large number of approximate density functionals and basis sets to confirm its validity.It has been found that the validity of thecis-effect is more dependent on the quality of basis sets than on that of approximate density functionals.We have also used a few well-established analysis tools,such as natural bond orbital,energy decomposition analysis,density functional reactivity theory,and non-covalent interaction analysis,to pinpoint its origin.A weak non-covalent interaction between the two substituting groups in thecisconformer has been identified and illustrated in all the systems studied by NBO and NCI analyses,but this kind of interactions could be both attractive and repulsion and one cannot use it to account for the validity of thecis-effect.Employing the two energy decomposition schemes,we observed a rather complicated picture about the behaviors of different energy components,where no single interaction was found to be solely responsible for the validity of the effect.Nevertheless,we found that a reasonably good correlation between the electrostatic interaction and the total energy difference with the correlation coefficientR2equal to 0.71.If two variable least-square fits are used with the electrostatic interaction plus either the kinetic energy or DFT steric energy,better correlations were obtained withR2equal to 0.85.Thecisisomer was found to possess larger exchange-correlation interactions,less steric repulsion,and stronger fermionic quantum repulsion in almost all the cases studied,with the less steric repulsion result verified by the less molar volume.We anticipate that the conclusions drawn from the present study are applicable to other similar systems as well.
(1) (a)Craig,N.C.;Entemann,E.A.J.Am.Chem.Soc.1961,83,3047.doi:10.1021/ja01475a019
(b)Craig,N.C.;Overend,J.J.Chem.Phys.1969,51,1127.
(c)Craig,N.C.;Piper,L.G.;Wheeler,V.L.J.Phys.Chem.1971,76,1453.
(d)Craig,N.C.;Chen,A.;Suh,K.H.;Klee,S.;Mellau,G.;Winnewiser,B.P.;Winnewisser,M.J.Phys.Chem.A1997,101,9302.
(e)Craig,N.C.;Brandon,D.W.;Stone,S.C.;Lafferty,W.J.J.Phys.Chem.1992,96,1598.
(2) Craig,N.C.;Lo,Y.S.;Piper,L.G.;Wheeler,J.C.J.Phys.Chem.1970,74,1712.doi:10.1021/j100703a011
(3) (a)Wood,R.E.;Stevenson,D.P.J.Am.Chem.Soc.1941,63,1650.doi:10.1021/ja01851a042
(b)Gardner,D.V.;McGreer,D.E.Can.J.Chem.1970,48,2104.
(4) (a)Salomma,P.;Nissi,P.Acta Chim.Scand.1967,21,1386.doi:10.3891/acta.chem.scand.21-1386
(b)Crump,J.W.J.Org.Chem.1963,28,953.
(c)Harwell,K.E.;Hatch,L.F.J.Am.Chem.Soc.1955,77,1682.
(5)Waldron,J.T.;Snyder,W.H.J.Am.Chem.Soc.1973,95,5491.doi:10.1021/ja00798a010
(6) Huber-Wälchli,P.;Günthard,H.H.Spectrochim.Acta1981,37,285.doi:10.1016/0584-8539(81)80159-6
(7) Durig,J.R.;Liu,J.;Little,T.S.;Kalasinsky,V.F.J.Phys.Chem.1992,96,8224.doi:10.1021/j100200a006
(8)Connor,T.M.;McLauchlan,K.A.J.Phys.Chem.1965,69,1888.doi:10.1021/j100890a018
(9) Epiotis,N.D.J.Am.Chem.Soc.1973,95,3087.doi:10.1021/ja00791a001
(10) Kollman,P.A.J.Am.Chem.Soc.1974,96,4363.doi:10.1021/ja00821a003
(11) Bemardi,F.;Bottoni,A.;Epiotis,N.D.;Guena,M.J.Am.Chem.Soc.1978,100,6018.doi:10.1021/ja00487a007
(12) (a)Cremer,D.J.Am.Chem.Soc.1981,103,3633.doi:10.1021/ja00403a003
(b)Cremer,D.Chem.Phys.Lett.1981,81,481.
(13) Carlos,J.L.;Karl,R.R.;Bauer,S.H.J.Chem.Soc.Faraday Trans.21974,2,177.
(14) Gandhi,S.R.;Benzel,M.A.;Dykstra,C.E.;Fukunaga,T.J.Phys.Chem.1982,86,3121.doi:10.1021/j100213a013
(15) Saebø,S.;Sellers,H.J.Phys.Chem.1988,92,4269.doi:10.1021/j100326a006
(16) Dixon,D.A.;Smart,B.E.;Fukunaga,T.Chem.Phys.Lett.1986,125,447.doi:10.1016/0009-2614(86)87076-2
(17)Yamamoto,T.;Kaneno,D.;Tomoda,S.Chem.Lett.2005,34,1190.doi:10.1246/cl.2005.1190
(18) Parr,R.G.;Yang,W.Density-Functional Theory ofAtoms and Molecules.InInternationalSeriesofMonographs onChemistry;Clarendon Press:Oxford,England,1989;Vol.16,p 333.
(19) Liu,S.B.J.Chem.Phys.2007,126,244103.doi:10.1063/1.2747247
(20) Liu,S.B.;Govind,N.;Pedersen,L.G.J.Chem.Phys.2008,129,094104.doi:10.1063/1.2976767
(21) Liu,S.B.;Hu,H.;Pedersen,L.G.J.Phys.Chem.A2010,114,5913.doi:10.1021/jp101329f
(22) Ess,D.H.;Liu,S.B.;DeProft,F.J.Phys.Chem.A2010,114,12952.doi:10.1021/jp108577g
(23) Tsirelson,V.G.;Stash,A.I.;Liu,S.B.J.Chem.Phys.2010,133,114110.doi:10.1063/1.3492377
(24)Huang,Y.;Zhong,A.G.;Yang,Q.S.;Liu,S.B.J.Chem.Phys.2011,134,084103.doi:10.1063/1.3555760
(25) Torrent-Sucarrat,M.;Liu,S.B.;DeProft,F.J.Phys.Chem.A2009,113,3698.doi:10.1021/jp8096583
(26) Hohenberg,P.;Kohn,W.Phys.Rev.B1964,136,864.doi:10.1103/PhysRev.136.B864
(27) Becke,A.D.Modern Electronic Structure Theory;Yarkony,D.R.Ed.;World Scientific:River Edge,N.J.,1995;pp 1022-1046.
(28) Cohen,A.J.;Mori-Sánchez,P.;Yang,W.Science2008,321,792.doi:10.1126/science.1158722
(29)Xu,H.Y.;Wang,W.Acta Phys.-Chim.Sin.2011,27,2565.[许惠英,王 维.物理化学学报,2011,27,2565.]doi:10.3866/PKU.WHXB20111127
(30)Bader,R.F.W.;Essén,H.J.Chem.Phys.1984,80,1943.doi:10.1063/1.446956
(31) Johnson,E.R.;Keinan,S.;Mori-Sánchez,P.;Contreras-García,J.;Cohen,A.J.;Yang,W.J.Am.Chem.Soc.2010,132,6498.doi:10.1021/ja100936w
(32) Geerlings,P.;DeProft,F.;Langenaeker,W.Chem.Rev.2003,103,1793.doi:10.1021/cr990029p
(33) Liu,S.B.Acta Phys.-Chim.Sin.2009,25,590.[刘述斌.物理化学学报,2009,25,590.]doi:10.3866/PKU.WHXB20090332
(34) Woon,D.E.;Dunning,T.H.,Jr.J.Chem.Phys.1993,98,1358.doi:10.1063/1.464303
(35) Dunning,T.H.,Jr.;Hay,P.J.Modern Theoretical Chemistry;Schaefer,H.F.,III.Ed.;Plenum:New York,1976;Vol.3,pp 1-28.
(36) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian-09,Revision B.01;Gaussian Inc.:Wallingford,CT,2009.
(37) NBO Version 3.1,Glendening,E.D.;Reed,A.E.;Carpenter,J.E.;Weinhold,F.doi:10.3878/j.issn.1006-9585.2012.11212
(38) Valiev,M.;Bylaska,E.J.;Govind,N.;Kowalski,K.;Straatsma,T.P.;van Dam,H.J.J.;Wang,D.;Nieplocha,J.;Apra,E.;Windus,T.L.;De Jong,W.A.Comput.Phys.Commun.2010,181,1477.doi:10.1016/j.cpc.2010.04.018
(39) Dunning,T.H.,Jr.J.Chem.Phys.1989,90,1007.doi:10.1063/1.456153
(40) NBO Version 5.0,Glendening,E.D.;Badenhoop,J.K.;Reed,A.E.;Carpenter,J.E.;Bohmann,J.A.;Morales,C.M.;Weinhold,F.(Theoretical Chemistry Institute,University of Wisconsin,Madison,WI,2001).http://www.chem.wisc.edu/-nbo5.
(41) Weinhold,F.;Landis,C.Valency and Bonding:A Natural Bond Orbital Donor-Acceptor Perspective;Cambridge University Press:UK,2005.
(42) Contreras-García,J.;Johnson,E.R.;Keinan,S.;Chaudret,R.;Piquemal,J.P.;Beratan,D.N.;Yang,W.J.Chem.Theory Comput.2011,7,625.doi:10.1021/ct100641a
(43) Humphrey,W.;Dalke,A.;Schulten,K.J.Mol.Graphics1996,14,133.
(44)AIMAll(Version 11.08.23),Keith,T.A.TK Gristmill Software,Overland Park KS,USA,2012(aim.tkgristmill.com);Bader,R.F.W.AtomsinMolecules:AQuantumTheory;OxfordUniversity Press:Oxford,1990;Popeplier,P.L.;Hall,P.AtomsinMolecules:An Introduction;London,2000;Matta,C.F.,Boyd,R.J.Eds.;The Quantum Theory of Atoms in Molecules:From Solid Stateto DNA and Drug Design;Wiley:Weinham,2007.
(45) Feller,D.;Peterson,K.A.;Dixon,D.A.J.Phys.Chem.A2011,115,1440.
(46) (a)Liu,S.B.Phys.Rev.A1996,54,1328.doi:10.1103/Phys RevA.54.1328
(b)Liu,S.B.;Parr,R.G.Phys.Rev.A1996,53,2211.
(c)Nagy,A.;Liu,S.B.;Parr,R.G.Phys.Rev.A1999,59,3349.
(d)Liu,S.B.;Morrison,R.C.;Parr,R.G.J.Chem.Phys.2006,125,174109.
(47) (a)Liu,S.B.;Pedersen,L.G.J.Phys.Chem.A2009,113,3648.doi:10.1021/jp811250r
(b)Liu,S.B.;Schauer,C.K.;Pedersen,L.G.J.Chem.Phys.2009,131,164107.
(c)Burger,S.K.;Liu,S.B.;Ayers,P.W.J.Phys.Chem.A2011,115,1293.
(d)Huang,Y.;Liu,L.;Liu,W.;Liu,S.G.;Liu,S.B.J.Phys.Chem.A2011,115,14697.
(e)Huang,Y.;Liu,L.;Liu,S.B.Chem.Phys.Lett.2012,527,73.
猜你喜欢
北京航空航天大学学报(2021年9期)2021-11-02
北京航空航天大学学报(2021年7期)2021-08-13
北京航空航天大学学报(2021年6期)2021-07-20
泰山学院学报(2019年6期)2020-01-14
山西教育·招考(2019年12期)2019-09-10
山西教育·招考(2019年12期)2019-09-10
山西教育·招考(2019年12期)2019-09-10
物理化学学报(2018年6期)2018-03-08
药学研究(2015年11期)2015-12-19
天津商务职业学院学报(2015年2期)2015-02-28

- 物理化学学报的其它文章
- Thermodynamic Properties of Nd(Gly)2Cl3·3H2O and Pr(Ala)3Cl3·3H2O
- Vapor-Liquid Equilibrium Data for Carbon Dioxide+Dimethyl Carbonate Binary System
- lmproving Photocatalytic Performance for Hydrogen Generation over Co-Doped Znln2S4under Visible Light
- Core-Shell Nanospheres(HP-Fe2O3@TiO2)with Hierarchical Porous Structures and Photocatalytic Properties
- 中国化学会第四届全国热分析动力学与热动力学学术会议(第一轮通知)
- 第十一届东方胶化杯全国胶体化学研究生优秀成果奖评选通知