
Bromodomain小分子抑制剂的研究进展
2013-09-11 01:41王功金向华
当代化工研究 2013年5期
王功金 向华
(b 中国药科大学江苏省药物分子设计与成药性优化重点实验室,南京,210009)
Bromodomain小分子抑制剂的研究进展
王功金1a向华2*,a,b
(b中国药科大学江苏省药物分子设计与成药性优化重点实验室,南京,210009)
草甘膦是一种高效、低毒的灭生性除草剂,在世界范围内得到广泛应用。本文概述了IDA法和甘氨酸法草甘膦母液的主要成分,总结了草甘膦的主要回收方法,指出了现有研究的不足之处,为进一步研究提供参考。
草甘膦 除草剂 母液 回收方法
近年来的研究发现,组蛋白的翻译后修饰(Posttranslational modifications, PTMs)可通过影响DNA键合、蛋白质之间的相互作用调节一系列的细胞生理过程在基因的表达中发挥关键作用。这一过程通过“写—读—擦” (Write-Read-Erase)机制进行[1,2,3]。组蛋白的翻译后修饰主要对组蛋白赖氨酸残基的N端裸露残基电荷进行中和,降低了对带负电的DNA的亲和性。其主要形式有乙酰化、甲酰化、磷酸化核糖基化、生物素化、瓜氨酸化、巴豆酰化和泛素蛋白化等。其中,对赖氨酸残基的乙酰化作用(ε-N-acetylation of lysine residues, Kac)是最显著的翻译后修饰方式[1,4]。研究表明,组蛋白的乙酰化作用使其作为组合密码调控基因表达,影响基因表达产物并传递给下游分子,进而产生生化效应[5-10]。赖氨酸残基的乙酰化作用由组蛋白乙酰化酶(Histone aceyltransferase, HAT)(writers)催化生成,BRD蛋白可特异性的与赖氨酸乙酰化物结合从而识别赖氨酸乙酰化物(readers),生成后的乙酰基可由组蛋白去乙酰化
酶(Histone deactylases, HDAC)(erasers)水解脱去。目前,所有与组蛋白乙酰化酶有关的转录辅激活因子都包括Bromodomains (BRDs)[11],而BRDs也是目前已知的唯一能对赖氨酸乙酰化物特异性识别的蛋白[12]。鉴于翻译后修饰作用在基因表达中的重要作用,能阻断赖氨酸乙酰化物与BRDs特异性结合的小分子化合物近年来逐渐成为研究热点。
1 BRD蛋白家族
1.1 BRD蛋白
BRD蛋白是一种进化上高度保守的约含110个氨基酸的溴蛋白质功能结构域,通过染色质的组装和乙酰化而参与信号依赖性的、非基础性的基因转录调控。BRD蛋白亦可通过对转录因子等非组蛋白的乙酰化修饰而广泛参与细胞周期调控、细胞分化、信号转导等过程[13]。
BRD蛋白最开始在果蝇的基因中发现并被确证,并由此命名,后来在人和酵母的基因中也发现有BRD蛋白[14]。人类的基因组在42种不同的蛋白质中编码至少56种BRD蛋白。BRD蛋白广泛分布于组蛋白乙酰转移酶、转录共激活因子、核激酶等染色质相关蛋白质中,且大多在核内发挥转录因子的作用。BRD蛋白通常不单独存在,而是作为一个更大的含BRD蛋白(BRD containing proteins BCPs)的一部分。其在蛋白质中一般出现一次或两次,最多可出现五次。同一蛋白质中不同的BRD蛋白的功能可能并不相同,可能作用于不同的核内底物或影响蛋白的核定位[15,16,17]。
BRD家族共包含61种BRD蛋白,可根据其母蛋白的功能划分为以下8个亚型[11,17-27]。1. 组蛋白乙酰化酶类,如GCN5、PCAF等;2. Bromodomain and extraterminal (BET)家族,如BRD2、BRD3、BRD4和BRDT;3. ATP依赖的染色质重塑复合物的因子,如Swi2/Snf2、BAZ1B等;4. 解旋酶类,如SMARCA等;5. 甲基转移酶类,如MLL、ASH1L等;6. 转录辅激活因子类,如TRIM/TIF1等;7. 调节酶类,如TAF1等;8. 核骨架蛋白类,如PB1等。研究表明,大部分BRD蛋白都在调控如组蛋白乙酰化酶、依赖ATP的染色质重塑、甲基化转移酶和转录激活因子等基因转录过程中发挥重要作用[18,28],并与肿瘤、神经紊乱、炎症、肥胖和心血管疾病等疾病的发生密切相关[29,3031]。
1.2 BRD蛋白结构
Dhalluin C. 等于1999年用核磁共振手段首次完成对BRD蛋白结构的确定[11]。近年来,Filippakopoulos等通过核磁共振和X-ray晶体衍射技术对更多的BRD蛋白进行了结构确证[32],通过对其三维结构的研究发现它们具有共同的结构基础。
BRD蛋白呈一反平行排列左手方向的四螺旋丛(antiparallel left-handed four helibundle)。第一和第二、第三和第四螺旋之间形成两个疏水性的袢环结构,分别称ZA、BC环。该袢环结构能与赖氨酸乙酰化物结合使得BRD蛋白具有辨别不同蛋白结合物的能力[11,13,33]。BRD蛋白通过此袢环结构可特异性地与组蛋白N末端乙酰化赖氨酸位点相结合,传递组蛋白乙酰化信号、调控基因转录。赖氨酸乙酰化物可通过氢键的作用与最多由四个排列有序的结晶水形成基底的疏水口袋识别、结合。多晶体结构分析表明赖氨酸乙酰化物的识别氢键由赖氨酸乙酰化物的乙酰羰基上的氧原子与BRD蛋白分子内在结构上高度保守的天门冬氨酸、苏氨酸或酪氨酸上的氨基形成[12,13,34,35](图1)。
BRD蛋白是人体内唯一的特异性识别组蛋白赖氨酸残基乙酰化物的作用模型,在疾病治疗、药物研发等方面的研究价值不言而喻。因此,可设计小分子药物和BRD蛋白的疏水口袋结合从而达到抑制BRD蛋白特异性识别组蛋白赖氨酸乙酰化物的目的,进而治疗疾病。
2 BRD蛋白小分子抑制剂
2.1 1-N-芳基-1,3-丙二胺类[36]
最早发现的BRD蛋白小分子抑制剂1(图2)是以结构为基础、以核磁共振为手段,通过检测BRD蛋白与配体结合前后的2D 15N谱图的变化为原理筛选获得的。该类化合物被设计用于治疗HIV。传统的HIV药物因HIV病毒的快速变异而失去治疗活性,该类化合物的优势在于其靶蛋白是宿主细胞的PCAF BRD蛋白而不是病毒蛋白,使得其能最大程度地降低因病毒变异带来抗药性。其对PCAF BRD蛋白的IC50可达1.6μM。另外,该类化合物具有较高的选择性,其对与PCAF BRD蛋白结构相似的CBP和TIF1β蛋白在该浓度级别无结合活性。
通过IC50值的测定、3D结构与PCAF BRD蛋白的模拟对接,总结了该类化合物的构效关系(表1)。
由表1可见,1-N-苯环上4-甲基为必需基团,引入4-甲基可使亲和性明显提高,当4-甲基换为4-乙基、3-或5-甲基都使化合物分子与BRD蛋白的亲和性降低,4-苯基的取代物甚至丧失了亲和活性。4-或5-氰基取代物的亲合活性也很低。此外,1-N-苯环上的2-硝基和1位-脂肪长链末端的氨基也是该类化合物的必需基团,当末端氨基被换为羧基等基团时,其亲和活性消失。脂肪族长链以三个碳的长度时亲和性为最佳。当长链上有四个碳时,其亲和性下降。而当其长链上有两个碳时,甚至没有亲和活性。

图1 BRD蛋白结构及其和组蛋白乙酰化物作用模型[12]Figure 1 Overall structure of BRD and binding of Kac on the central cavity of module[12]
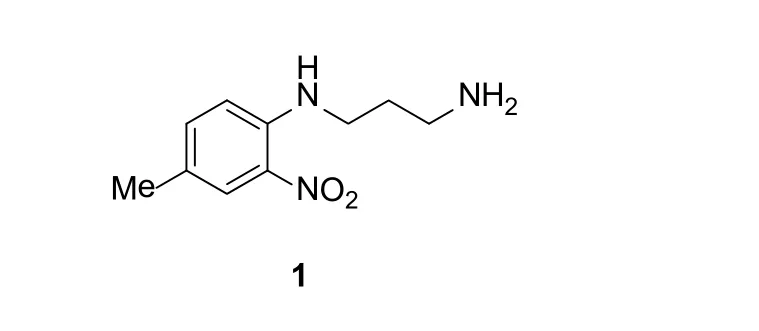
图2 PCAF BRD蛋白小分子抑制剂1Figure 2 Structure of the PCAF bromodomain inhibitor 1
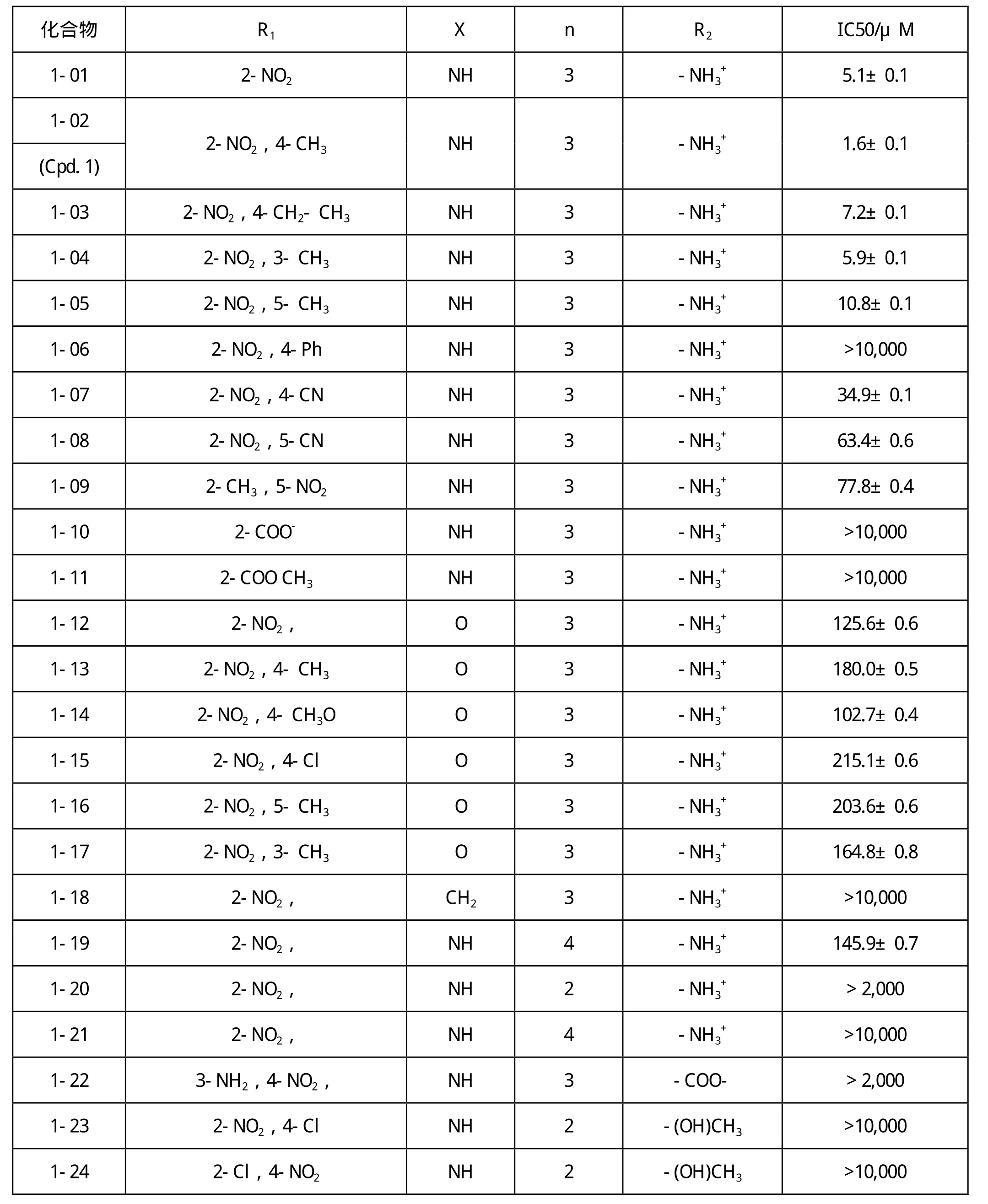
表1 化合物1及其衍生物的构效关系Table 1 Structure-Activity Relationship for Compound 1 and Its Derivatives
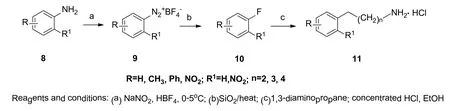
图3 1-N-芳基-1,3-丙二胺类的合成Figure 3 Synthesis of N-1-aryl-propane-1, 3-diamine compounds
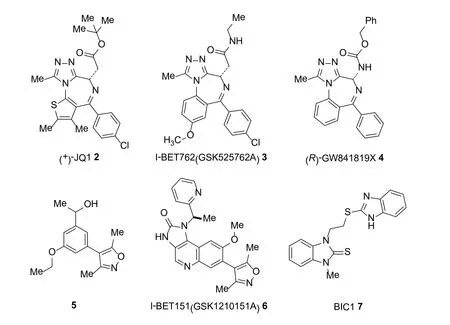
图4 BET蛋白选择性抑制剂Figure 4 Structures of the BET bromodomain-selective inhibitors
1-N-芳基-1,3-丙二胺类化合物合成方法如图3所示。
该类化合物和PCAF BRD蛋白结合物的核磁结构分析表明结合位点位于BRD蛋白ZA环的E756残基和αB螺旋末端的Y802之间。甲基基团和ZA环内的小疏水口袋结合,进一步促进了该类化合物和PCAF BRD蛋白家族的亲和性[37]。
虽然对该类化合物的结构优化工作仍在继续,但目前未见更高亲和性的分子结构报道[38]。
2.2 BET家族的选择性抑制剂
2.2.1 BET家族
近年来,因BET蛋白家族在抗肿瘤、抗感染、抗病毒等方面的潜在价值,使得在开发小分子BRD蛋白抑制剂方面,药物化学家们的注意力主要集中在BET蛋白家族选择性抑制剂范围内[29,39,40]。
BET家族是BRD蛋白家族的一部分。它们与其他的BRD蛋白的不同之处在于它们有两个在序列上高度保守的N端和一个C端结构域。研究表明,BRD4蛋白在基因表达的M、G1期结合于转录的开始位点,影响有丝分裂进程[41,42]。BRD4蛋白同时也是转录延伸的关键性介质,用于吸附正电性的转录延伸因子(P-TEFb)[43,44]。细胞周期蛋白激酶9是转录延伸因子(P-TEFb)的核心部分。它是治疗慢性淋巴细胞性白血病的一个常用靶点[45-48]。BRD4将转录延伸因子吸附至有丝分裂染色体上,从而促进了促生长基因的表达[41,49]。同时,组合实验证明,BRD4蛋白还参与HIV、EBV、HPV等病毒的转录调控[50,51,52]。另外,BRDT蛋白在不育的雄性老鼠的生殖细胞分裂中扮演重要角色[53]。
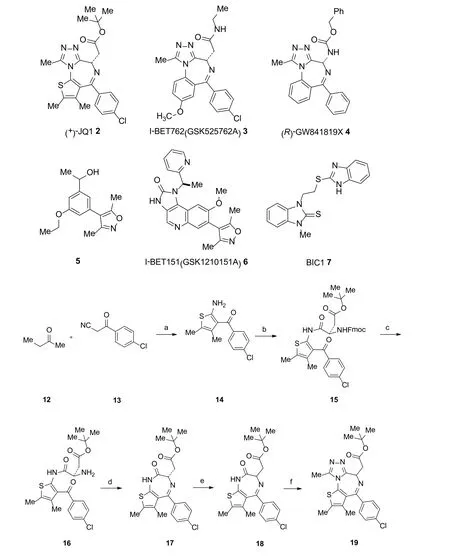
图5 2的外消旋体的合成Figure 5 Synthesis of Racemic 2
目前,有四类化学结构类型截然不同的小分子化合物能选择性地抑制BET蛋白和组蛋白乙酰化物的结合。它们分别是:甲基三氮唑并二氮杂类、3,5-二甲基异噁唑类、苯并咪唑类和苯并咪唑类 (图4)。
2.2.2 甲基三氮唑并二氮杂 类[54]
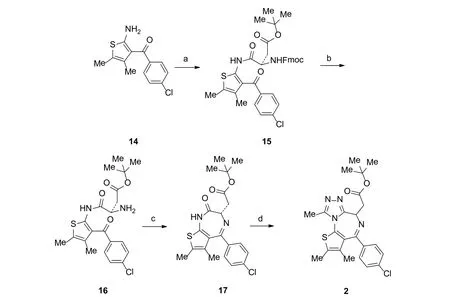
图6 (+)JQ1的合成Figure 6 Synthesis of (+)JQ1
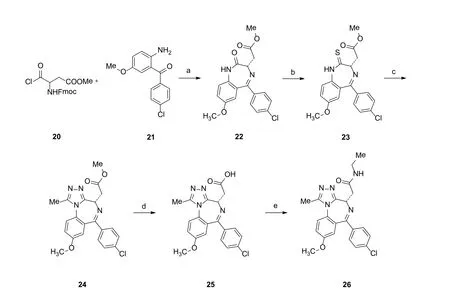
图7 I-BET762的合成Figure 7 Synthesis of I-BET762
化合物(+)JQ1(2)最初由高通量筛选得到,Filippakopoulos等首次化学合成JQ1的外消旋体(图5)及(+)JQ1(图6)。从已探明的该类化合物的构效关系来看,(+)JQ1结构中有位阻较大的叔丁基官能团,能有效地降低其与其他苯二氮 受体的亲和性。通过共晶结构技术可知,三唑环和BET蛋白赖氨酸残基末端进化上高度保守的精氨酸形成氢键,其与组蛋白乙酰化物的结合位点在物理形态上有极高的重合性,从而使得其占据了整个BRD蛋白的疏水口袋,稳定了亲和结构,进而高效地阻断组蛋白乙酰化物对BRD蛋白的特异性结合。而其光学异构体(-)-JQ1因二氮杂 环的扭曲而和BRD蛋白ZA-loop区域的空间位阻使得其与目前所有的BRD蛋白都没有显著的亲和性。通过荧光测定法(DSF)及恒温热量滴定(ITC)等方法测定,光学纯(+)-JQ1与BRD4蛋白的亲和半抑制浓度值可达50nM,显示其在肿瘤及异种移植等领域的潜在价值。(+)JQ1 对BET蛋白具有较高的选择性,而对其他的BRD蛋白,如WDR9(2)和CREBBP等没有显著的亲和活性。
Nicodeme[55]和Chung[56]等通过表型筛选的方法得到化合物3。该化合物被设计用来研究载脂蛋白A1的小分子正性调节物,但实验结果指出该化合物并未达到上述目的。当他们把该类化合物和HepG2细胞接触后发现BET蛋白家族的BRD2、BRD3、BRD4可能是这类化合物的细胞靶点。实验表明,3和4都能阻断四乙酰化的组蛋白肽和BET家族的BRD2、BRD3、BRD4等的相互作用,对BET家族外的BRD蛋白却不显示任何亲和活性。
另外,最近的研究表明,儿童与青年睾丸核蛋白中线癌(nuclear protein in testis midline carcinoma,NMC)是一种完全由基因特征来定义的疾病,由BRD4-NUT基因易位引起。这种由两个来自不同染色体的基因连接在一起形成的异常的合并蛋白被称为BRD4-NUT[54]。3被GSK用来治疗NMC的开发已处于临床研究阶段[13]。
Filippakopoulos等通过对该类化合物的构效关系的研究也给出了27和28(图8)这类苯二氮 类虽可以和BET蛋白结合,但没有微摩尔活性的原因。镇静咪达唑仑28分子内是咪唑环而非该类化合物的三氮唑环,因而不能和天冬酰胺残基乙酰化物形成氢键。虽然其能与BET蛋白结合,但弱于三氮唑,而使得在常规剂量下,起不到阻断BET识别组蛋白乙酰化物的作用。虽然,咪唑环更具有柔性,但在乙酰化物的结合位点,甲基取代基是亲和活性的必需基团,当把甲基取代基换为氢乙基、苯基、氨基或羰基都会降低其与BET蛋白的亲和活性[57]。
2.2.3 3,5-二甲基异噁唑类
近年来,Hewings[58],Dawson[59]和Bamorough[60]等的最新研究表明,3,5-二甲基异噁唑类化合物作为乙酰化物的类似物是有效的BET蛋白选择性抑制剂。Hewing等在研究中发现,N-甲基吡咯烷酮对一系列的BRD蛋白显示轻微的亲和活性。在对该类含甲基杂环的研究中发现含二氢喹啉酮的3,5-二甲基异噁唑结构对BRD2和BRD4的IC50分别为3μM和7μM,进而发现含有3,5-二甲基异噁唑结构的29(图9)能占据赖氨酸乙酰化物对BRD4的结合位点,并以此结构为指导设计出更小分子量的含有4-苯基-3,5-二甲基异噁唑结构的化合物5[58]。该化合物对BET蛋白家族有良好的选择性。5和BRD4的结合物的X-ray晶体衍射显示,结合方式与3和4相似。3,5-二甲基异噁唑类部分的氧原子和N140的胺基形成氢键,N原子和Y97中的结合水形成氢键。乙氧取代基和ZA环相邻却不和其中的结合水形成氢键。这些氢键符合理论的氮原子和氧原子单独形成强氢键之间的角度,若将氮原子和氧原子的排列颠倒则就会形成较弱的氢键[37]。据报道,5对BRD4的亲和性的半抑制浓度值可达5μM[58]。
为了进一步提高该类化合物的亲和活性,Bamborough等人通过封装晶体的方法观察氢键和疏水性的相互作用,构建了一个三维药效团模型。以这个模型筛为标准,筛选出一系列带有连在苯环上的硫胺的4-苯基-3,5-二甲基异噁唑类似物(35-40)(图11)。
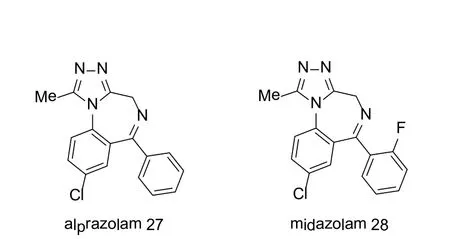
图8 阿普唑仑和咪达唑仑Figure 8 Structures of Alprazolam and Midazolam
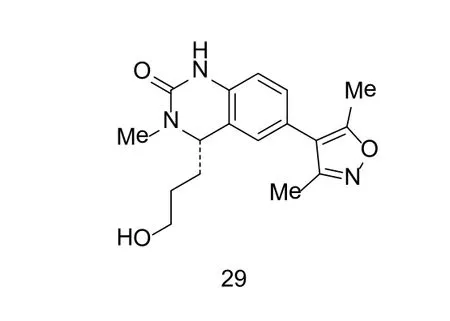
图9 先导化合物29Figure 9 The lead compound 29
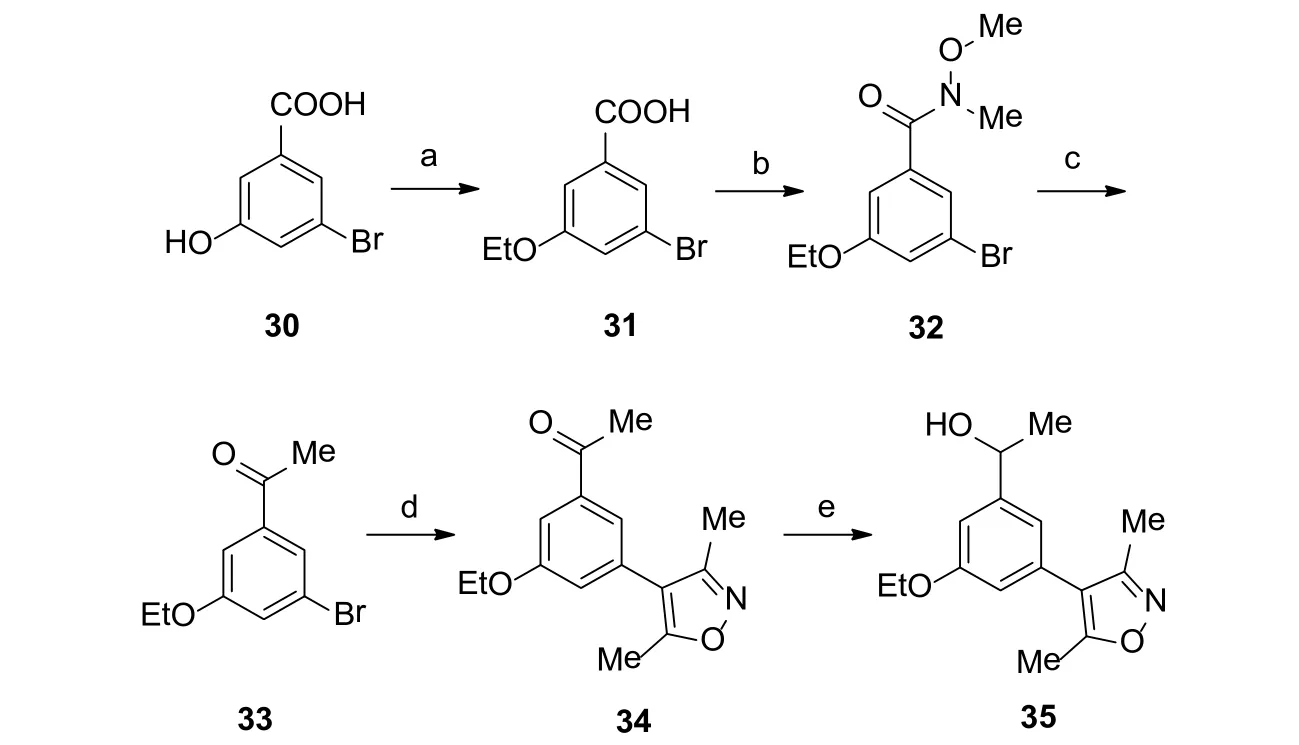
图10 化合物5的合成[58]Figure 10 Synthesis of 5[58]
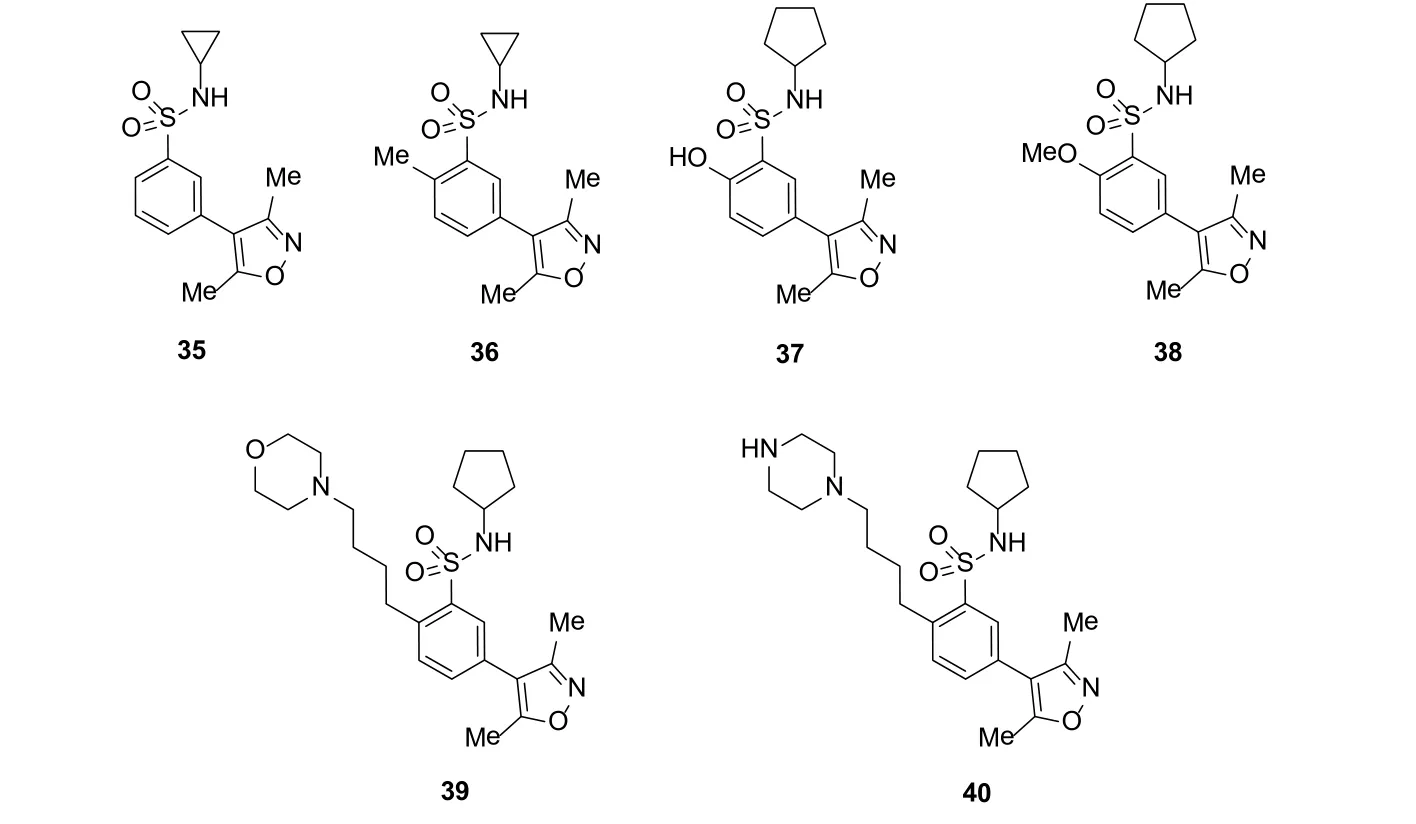
图11 Bamborough等筛选异噁唑类似物结构Figure 11 Structures of the isoxazoles selected by Bamborough etc.
这些化合物除了基本结构外,硫胺结构也可以和水形成一定数量的氢键从而增加亲和性。对该类化合物的研究表明,化合物38的活性最好。通过热转移结合分析发现,其对BET蛋白有特殊的选择性。值得指出的是,化合物38还显示一定的细胞活性。然而,该类化合物受到其溶解度的限制,因此,又设计出一类在该类化合物的苯环上添加增溶性基团的类似物。虽然这种修饰从一定程度上减少了其亲和活性,却增加了耐受性和水溶性[37]。
该系列化合物的合成路线如图12所示。
6的发现和3相似。该类化合物中首先被设计作为载脂蛋白A1的小分子正调节物[61],而后才发现其和BRD蛋白的亲和活性。
对该类化合物的构效关系研究表明,1,4位-氨基上连接一系列疏水基团后,能表现出良好的耐受性;将3位的酰胺环化形成脲也能表现出良好的耐受性,却有意外的环磷酰胺抑制;但使用适当的胺基取代基或在6位添加一个甲基取代基都能破坏这些脲的环磷酰胺抑制;3, 6位在亲和性、配体取代能力和抑制细胞因子释放等方面和3的活性相当,但在药物动力学方面比2和3更加优越。小鼠实验显示,6的半衰期为7.2h,而2和3接近,都为1.6h左右。[59,62]
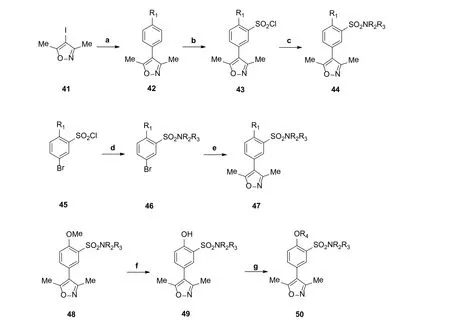
图12 Bamborough等筛选异噁唑类似物的合成[60]Figure 12 Synthesis of the isoxazoles selected by Bamborough etc.[60]
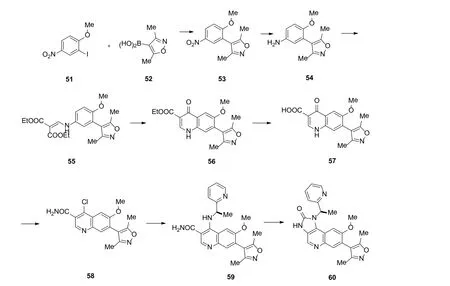
图13 6的合成[59]Figure 13 Synthesis of 6[59]
2.2.4 苯并咪唑类
Ito[63]等人发现以苯为基础的BET抑制剂7。该类化合物阻断BRD2和组蛋白乙酰化物之间的结合作用。X射线晶体衍射显示,苯并咪唑的苯环部分占据了乙酰化物的结合口袋,而2-硫酮苯并咪唑部分这和WPF架直接作用。该类化合物并不和在结构上高度保守的Asn和Tyr残基形成氢键,从一定程度上解释了该类化合物对BRD2的亲和活性较低的原因。
3 展望
表观遗传学最早由Waddington等于1939年提出[64],而于近年来发展成为药物研究的热点[65]。它可以理解为在不涉及DNA列序变化的前提下,发生于细胞分裂中有关基因的表达和表型的遗传变化等。有证据表明,组蛋白的翻译后修饰和DNA的修饰一样,都可以通过复杂的细胞循环修复。因此,组蛋白翻译后修饰有望成为一种表观遗传方法[65-69]。有理由相信, BRD蛋白的选择性小分子抑制剂将成为研究BRD蛋白的细胞功能和相关疾病治疗的重要工具。随着基础研究的继续深入,越来越多的选择性和生物活性更加优良的BRD蛋白小分子抑制剂将为以表观遗传域为靶点的治疗方案提供新的治疗手段。
[1]Kouzarides, T. Chromatin modifications and their function. Cell 2007,128, 693-705.
[2]Shogren-Knaak, M.; Ishii, H.; Sun, J. M.; et al. Histone H4-K16 acetylation controls chromatin structure and pro-tein interactions.Science 2006, 311, 844-847.
[3]Celic, I.; Masumoto, H.; Griffith, W. P.; et al. The sirtuins hst3 and Hst4p preserve genome integrity by controlling histone h3 lysine 56 deacetylation. Curr. Biol. 2006, 16, 1280-1289.
[4]Turner, B. M. Reading signals on the nucleosome with a new nomenclature for modified histones. Nat. Struct. Mol. Biol. 2005, 12,110-112.
[5]Winston, F.; Allis, C. D. The bromodomain: a chromatin-targeting module? Nat. Struct. Biol. 1999, 6, 601-604.
[6]Strahl, B. D.; Allis, C. D. The language of covalent histone modifications.Nature 2000, 403, 41-45.
[7]Jenuwein, T.; Allis, C. D. Translating the histone code. Science 2001,293, 1074-1080.
[8]Agalioti, T.; Chen, G.; Thanos, D. Deciphering the transcriptional histone acetylation code for a human gene. Cell 2002, 111, 381-392.
[9]Loyola, A.; Almouzni, G. Bromodomains in living cells participate in deciphering the histone code. Trends Cell Biol. 2004, 14, 279-281.
[10]Gardner, K. E.; Allis, C. D.; Strahl, B. D. Operating on chromatin, a colorful language where context matters. J. Mol. Biol. 2011, 409, 36-46.
[11]Dhalluin, C.; Carlson, J. E.; Zeng, L.; et al. Structure and ligand of a histone acetyltransferase bromodomain. Na-ture 1999, 399, 491-496.
[12]Filippakopoulos P.; Knapp S. The bromodomain interaction module.FEBS letters 2012, 586, 2692-2704.
[13]Conway, S. J. Bromodomains: Are Readers Right for Epigenetic Therapy? ACS Medicinal Chemistry Letters 2012, 3, 691-694.
[14]Haynes, S. R.; Dollard, C.; Winston, F.; et al. The bromodomain: A conserved sequence found in human, Droso-phila and yeast proteins.Nucleic Acids Res. 1992, 20, 2603.
[15]Mark, H.; Sally, R.; Louis, C. acetyllysine-binding and function of bromodomain-containing proteins in chro-matine. Frontiers in Bioscience 2001, 6, 853-865.
[16]Francois, J.; Jean-Marie, W.; Bertrand, L. D. The bromodomain revisited. TIBS. 1997, 22, 151-153.
[17]Jacobson, R. H.; Ladurner, A. G.; King, D. S.; et al. Structure and function of a human TAFII250 double bromo-domain module. Science 2000, 288, 1422-1425.
[18]Muller, S.; Filippakopoulos, P.; Knapp, S. Bromodomains as therapeutic targets. Expert Rev. Mol. Med. 2011, 13, e29.
[19]Florence, B.; Faller, D. V. You bet-cha: a novel family of transcriptional regulators. Front. Biosci. 2001, 6, D1008-1018.
[20]Yang, X. J.; Ogryzko, V. V.; Nishikawa, J.; Howard, B. H.; Nakatani,Y. A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature 1996, 382, 319-324.
[21]Cavellan, E.; Asp, P.; Percipalle, P. The WSTF-SNF2h chromatin remodeling complex interacts with several nu-clear proteins in transcription. J. Biol. Chem. 2006, 281, 16264-16271.
[22]Trotter, K. W.; Archer, T. K. The BRG1 transcriptional coregulator.Nucl. Recept. Signal 2008, 6, e004
[23]Malik, S.; Bhaumik, S. R. Mixed lineage leukemia: histone H3 lysine 4 methyltransferases from yeast to human. FEBS J. 2010, 277, 1805-1821.
[24]Gregory, G. D.; Vakoc, C. R.; Rozovskaia, T.; et al. Mammalian ASH1L is a histone methyltransferase that occu-pies the transcribed region of active genes. Mol. Cell. Biol. 2007, 27, 8466-8479.
[25]Venturini, L. TIF1gamma, a novel member of the transcriptional intermediary factor 1 family. Oncogene 1999, 18, 1209-1217.
[26]Xue, Y. The human SWI/SNF-B chromatin-remodeling complex is r elated to yeast rsc and localizes at kinetocho-res of mitotic chromosomes. Proc. Natl. Acad. Sci. USA. 2000, 97, 13015-13020.
[27]Bres, V.; Yoh, S. M.; Jones, K. A. The multi-tasking P-TEFb complex.Curr. Opin. Cell Biol. 2008, 20, 334-340.
[28]Mujtaba, S.; Zeng, L.; Zhou, M. M. Structure and acetyl-lysine recognition of the bromodomain. Oncogene 2007, 26, 5521-5527.
[29]Chung, C-W.; Tough, D. F. Bromodomains: A New Target Class f or Small Molecule Drug Discovery, Drug Discovery Today Therapeutic Strategies 2012, 9, e111-e20.
[30]Denis, G. V. Bromodomain Coactivators in Cancer, Obesity, Type 2 Diabetes, and Inflammation. Discov. Med. 2010, 10, 489-499.
[31]Sanchez, R.; Zhou, M. M. The Role of Human Bromodomains in Chromatin Biology and Gene Transcription. Curr. Opin. Drug Discov.2009, 12, 659-665.
[32]Filippakopoulos, P.; Picaud, S.; Mangos, M.; et al. Histone recognition and large-scale structural analysis of the human bromodomain family.Cell 2012, 149, 214-231.
[33]Vollmuth, F.; Blankenfeldt, W.; Geyer, M. Structures of the dual bromodomains of the P-TEFb-activating protein Brd4 at atomic resolution. J. Biol. Chem. 2009, 284, 36547-36556.
[34]Vidler, L. R.; Brown, N.; Knapp, S. Druggability Analysis and Structural Classification of Bromodomain Ace-tyl-lysine Binding Sites. J. Med.Chem. 2012, 55, 7346-7359
[35]Bamborough, P.; Diallo, H.; Goodacre, J. D.; et al. Fragment-based discovery of bromodomain inhibitors part 2: optimization of phenylisoxazole sulfonamides. J. Med. Chem. 2012, 55, 587-596.
[36]Zeng, L.; Li, J.; Muller, M. et al. Selective small molecules blocking HIV-1 Tat and coactivator PCAF association. J. Am. Chem. Soc. 2005,127, 2376-2377.
[37]Hewings, D. S.; Rooney T. P.; Jennings, L. E.; et al. Progress in the Development and Application of Small Molecule Inhibitors of Bromodomain-Acetyl-Lysine Interactions. J Med Chem 2012, 55,9393-9413.
[38]Pan, C.; Mezei, M.; Mujtaba, S.; et al. Structure-guided optimization of small molecules inhibiting human immu-nodeficiency virus 1 Tat association with the human coactivator p300/CREB binding proteinassociated factor. J. Med. Chem. 2007, 50, 2285-2288.
[39]Muller, S.; Filippakopoulos, P.; Knapp, S. Bromodomains as therapeutic targets. Expert Rev. Mol. Med. 2011, 13, e29.
[40]Prinjha, R. K.; Witherington, J.; Lee, K. Place your BETs: the therapeutic potential of bromodomains. Trends Pharmacol. Sci. 2012,33, 146-153.
[41]Yang, Z.; He, N.; Zhou, Q. Brd4 recruits P-TEFb to chromosomes at late mitosis to promote G1 gene expression and cell cycle progression.Mol. Cell. Biol. 2008, 28, 967-976.
[42]Dey, A.; Nishiyama, A.; Karpova, T.; et al. Brd4 marks select genes on mitotic chromatin and directs postmitotic transcription. Mol Biol Cell 2009, 20, 4899-4909.
[43]Yang, X. J. Multisite protein modification and intramolecular signaling.Oncogene 2005, 24, 1653-1662.
[44]Yang, Z.; Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 2005, 19, 535-545.
[45]Peng, J.; Zhu, Y.; Milton, J. T. Identification of multiple cyclin subunits of human P-TEFb. Genes Dev. 1998, 12, 755-762.
[46]Marshall, N. F.; Price, D. H. Purification of P-TEFb, a transcription factor required for the transition into produc-tive elongation. J Biol Chem. 1995, 270, 12335-12338.
[47]Marshall, N. F.; Peng, J.; Xie, Z. Control of RNA polymerase II elongation potential by a novel carboxyl-terminal domain kinase. J Biol Chem. 1996, 271, 27176-27183.
[48]Phelps, M. A. Clinical response and pharmacokinetics from a phase 1 study of an active dosing schedule of fla-vopiridol in relapsed chronic lymphocytic leukemia. Blood 2009, 113, 2637-2645.
[49]Rahl, P. B.; Lin, Y. C.; Seila, A. C. c-Myc Regulates Transcriptional Pause Release. Cell 2010, 141, 4323-4445.
[50]Zhou, M.; Huang, K.; Jung, K. J.; et al. Bromodomain protein Brd4 regulates human immunodeficiency virus transcription through phosphorylation of CDK9 at threonine 29. J. Virol. 2009, 83, 1036-1044.
[51]Lin, A.; Wang, S.; Nguyen, T.; et al. The EBNA1 protein of Epstein-Barr virus functionally interacts with Brd4. J. Virol. 2008, 82, 12009-12019.
[52]Gagnon, D.; Joubert, S.; Senechal, H.; et al. Proteasomal degradation of the papillomavirus E2 protein is inhibited by overexpression of bromodomain-containing protein 4. J. Virol. 2009, 83, 4127-4139.
[53]Shang, E.; Nickerson, H. D.; Wen, D.; et al. The first bromodomain of Brdt, a testis-specific member of the BET sub-family of doublebromodomain-containing proteins, is essential for male germ cell differentiation. Development 2007, 134, 3507-3515.
[54]Filippakopoulos, P.; Qi J.; Picaud S.; et al. Selective Inhibition of Bet Bromodomains. Nature 2010, 468, 1067-1073.
[55]Nicodeme, E.; Jeffrey, K. L.; Schaefer, U.; et al. Suppression of inflammation by a synthetic histone mimic. Na-ture 2010, 468, 1119-1123.
[56]Chung, C.-W.; Coste, H.; White, J. H.; et al. Discovery and characterization of small molecule inhibitors of the BET family bromodomains. J. Med. Chem. 2011, 54, 3827-3838.
[57]Filippakopoulos, P.; Picaud, S.; Fedorov, O.; et al. Benzodiazepines and benzotriazepines as protein interaction inhibitors targeting bromodomains of the BET family. Bioorg. Med. Chem. 2012, 20, 1878-1886.
[58]Hewings, D. S.; Wang, M.; Philpott, M.; et al. 3,5-Dimethylisoxazoles act as acetyl-lysine-mimetic bromodomain ligands. J. Med. Chem.2011, 54, 6761-6770.
[59]Dawson, M. A.; Prinjha, R. K.; Dittmann, A.; et al. Inhibition of BET recruitmentto chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529-533.
[60]Bamborough, P.; Diallo, H.; Goodacre, J. D.; et al. Fragment-based discovery of bromodomain inhibitors part 2: optimization of phenylisoxazole sulfonamides. J. Med. Chem. 2012, 55, 587-596.
[61]Mirguet, O.; Lamotte, Y.; Donche, F.; et al. From ApoA1 upregulation to BET family bromodomain inhibition: discovery of I-BET151. Bioorg.Med. Chem. Lett. 2012, 22, 2963-2967.
[62]Seal, J.; Lamotte, Y.; Donche, F.; et al. Identification of a novel series of BET family bromodomain inhibitors: binding mode and profile of IBET151 (GSK1210151A). Bioorg. Med. Chem. Lett. 2012, 22, 2968-2972.
[63]Ito, T.; Umehara, T.; Sasaki, K.; et al. Real-time imaging of histone h4k12-specific acetylation determines the modes of action of histone deacetylase and bromodomain inhibitors. Chem. Biol. 2011, 18, 495-507.
[64]Waddington, C. H. Preliminary notes on the development of the Wings in normal and mutant strains of Droso-phila. Proc. Natl. Acad. Sci.USA. 1939, 25, 299-307.
[65]Arrowsmith, C. H.; Bountra, C.; Fish, P. V.; et al. Epigenetic protein families: a new frontier for drug discovery. Nat. Rev. Drug Discovery 2012, 11, 384-400.
[66]Biel, M.; Wascholowski, V.; Giannis, A. Epigenetics-an epicenter of gene regulation: histones and his-tone-modifying enzymes. Angew.Chem., Int. Ed. 2005, 44, 3186-3216.
[67]Holliday, R. Epigenetics: a historical overview. Epigenetics 2006, 1, 7 6-80.
[68]Probst, A. V.; Dunleavy, E.; Almouzni, G. Epigenetic inheritance during the cell cycle. Nat. Rev. Mol. Cell Biol. 2009, 10, 192-206.
[69]Sippl, W.; Jung, M. Epigenetic drug discovery special issue. Bioorg.Med. Chem. 2011, 19, 3603-3604.
Progress in the Research of Small Molecule Inhibitors of Bromodomains
Wang, Gongjin1aXiang, Hua2*,a,b
(a Department of Medicinal Chemistry, School of Pharmacy, China Pharmaceutical University, 24 Tong Jia Xiang Road, Nanjing 210009, Jiangsu, P. R. China)(bJiangsu Key Laboratory of Drug Design and Optimization, China Pharmaceutical University, 24 Tong Jia Xiang Road, Nanjing 210009, Jiangsu, P. R. China)
ε-N-acetylation of lysine residues on histone tails is one of the most important post-translational modification states in the human proteome. Bromodomains (BRDs) are the only interaction modules that specifically recognize Kac. Histone aceyl-transferases, BRDs and Histone deactylases form a system of “write-read-erase”, which has been linked with the transfer of epigenetic information. Over the past decade, the great attention has been paid to the potent and selective small molecule inhibitors targeting at BRDs. Herein, this review is focused on the discoveries, modes of action, biological activities, synthetic methods and the SAR of the small molecular BRD inhibitors to date.
Bromodomains; Small molecule inhibitors;Mechanism; SAR; Synthetic methods.
Q813
A
T1672-8114(2013)05-001-12
王功金(1987-),男,硕士研究生,药物化学专业,主要从事药物合成和工艺研究
猜你喜欢
中国生物化学与分子生物学报(2022年8期)2022-09-08
世界热带农业信息(2019年9期)2019-01-05
山东农业科学(2017年5期)2017-06-05
上海农业学报(2017年3期)2017-04-10
数字技术与应用(2016年10期)2017-04-01
广东饲料(2016年3期)2016-12-01
中国组织化学与细胞化学杂志(2016年4期)2016-02-27
动物营养学报(2015年10期)2015-12-01
中国当代医药(2015年16期)2015-03-01
应用化工(2014年10期)2014-08-16
