
眼遗传病的分子诊断
2013-11-01 03:09吴继红
中国眼耳鼻喉科杂志 2013年6期
吴继红
很多原发性的眼科疾病是遗传性的,目前已知眼科疾病中与遗传相关的有600余种。临床上常见的有视网膜母细胞瘤(retinoblastoma,RB)、视网膜色素变性(retinitis pigmentosa,RP)、青光眼、黄斑变性、先天性白内障(congenital cataract,CC)、Leber先天性黑矇(Leber congenital amaurosis,LCA)、Leber遗传性视神经病变(Leber hereditary optic neuropathy,LHON)、家族性渗出性玻璃体视网膜病变(familial exudative vitreoretinopathy,FEVR)、高度近视、角膜营养不良、先天性无虹膜。此外,还有上睑下垂、斜视、小眼球及无眼球等。根据遗传方式可分为4种主要类型:常染色体显性遗传病、常染色体隐性遗传病、性染色体连锁遗传病和线粒体遗传病。眼遗传病的分子基因学表现多样[1-2]:有些是单基因遗传病,单个基因突变就足以致病,如RB,其致病基因为RB1;另外一些属多基因遗传病,由多个基因、多个环境因子以及这些因子相互作用而致病,如RP、CC、青光眼等。一些常用的眼遗传病致病基因网站见表1。

表1 常用的眼遗传病致病基因网站
大部分眼遗传病存在遗传异质性,而且通过对不同地域或民族之间的比较发现,很多眼遗传病致病基因的突变位点和表型有较大差异,这为眼遗传病的分子基因学研究带来了很大困难。近年来,随着基因分析技术的快速发展,很多与眼遗传病相关的致病基因或致病突变位点被相继报道,这些已知的致病突变位点可作为基因标记,用于眼遗传病的临床基因诊断、症状前诊断和产前诊断。
眼遗传病的及时有效诊断、治疗对改善患者的生活质量有着重要意义。分子诊断是诊断眼遗传病不可缺少的手段和方法之一,不仅可通过确定发病基因来加深对疾病分子机制的理解,还可提供多种有效信息,有助于临床诊断和治疗、预测患者的病情进展及预后、发现无症状的携带者及症状前诊断。然而,目前国内对眼遗传病的分子基因学研究较少,眼遗传病的分子诊断也才刚刚起步,尚未建立起较规范和完善的眼遗传病分子诊断体系。
在此,我们以RB、RP等临床上常见的眼遗传病为例,介绍眼遗传病基因标记、分子诊断的常用技术,并讨论如何建立眼遗传病分子诊断服务。
1 常见眼遗传病的基因标记
一些常见眼遗传病的致病基因和基因标记见表2。

表2 常见眼遗传病的致病基因和基因标记
1.1 RB RB是儿童中最常见的眼内恶性肿瘤,我国每年新病例约1000例,占全世界每年新病例的20%左右,其中30% ~40%是属于遗传性型。RB是单基因遗传病,多为常染色体显性遗传,致病基因为RB1[3]。RB发病需要2次基因突变或发生表观遗传学改变[4-5]。第1次基因突变可发生于体细胞,也可发生于亲代生殖细胞。如果第1次突变发生于亲代生殖细胞则是遗传性的,有早发、多发的特点,而且其他组织产生第二肿瘤的可能性较高[6]。
1.2 RP RP是一组以视网膜光感受器进行性退变为特征的异质性遗传病。临床症状包括夜盲、视野变窄,最终导致失明,发病率约为1/3500[7]。RP的遗传方式多样,15% ~20%的患者是常染色体显性遗传,20%~25%的患者是常染色体隐性遗传,10% ~15%的患者是X染色体连锁遗传,剩下的40% ~55%的患者是散发的[8]。大部分RP是单基因致病,但也有双基因致病的[9-10];还有报道线粒体DNA突变可能也与RP 相关[11]。
据报道[12],至少有40个基因座与非综合征RP相关,其中已发现的致病基因超过60个。常染色体显性遗传的RP患者中,超过25%的致病基因为Rhodopsin(RHO),6% ~8%的致病基因为RP1[13]。约70%的X染色体连锁遗传RP患者的致病基因为Retinitis pigmentosa GTPase regulator(RPGR)[14]。
1.3 青光眼 青光眼是以视杯扩大和视野缺损为特征的异质性遗传病,也是很常见的致盲性眼病。其中以原发性开角型青光眼(primary open-angle glaucoma,POAG)最为常见,约占50%[15],在40岁以上人群中的发病率约为2%[16]。早发性POAG是常染色体显性遗传病,发病早且症状较重[17]。原发性先天性青光眼(primary congenital glaucoma,PCG)是常染色体隐性遗传病,多发于3岁以内的婴儿,是另一种重症青光眼[18]。
青光眼的发病机制尚不清楚。研究[12]发现,有4个基因座与PCG相关,而至少有15个基因座与POAG相关,其中已定位致病基因的有 GLC1A(myocilin,MYOC)[19],GLC1E(optineurin,OPTN)[20],GLC1G(WD repeatdomain 36, WDR36)[21], GLC3A(cytochrome P4501B1,CYP1B1)[22],GLC3D(latent transforming growth factor β binding protein 2,LTBP2)[23-24]。据报道,2% ~4%的欧美POAG患者有MYOC 基因突变[25-26],而 1.1% ~1.8%的中国 POAG患者发现该基因突变[27-28]。约16.7%的遗传性POAG患者和12%的散发性POAG患者有OPTN基因突变[20],而1% ~1.6%的中国POAG患者中发现该基因突变[29]。超过5%的 POAG患者有 WDR36基因突变[21,30]。48%的法国 PCG患者中发现 CYP1B1基因突变[31],而只有20%的日本患者中存在该基因突变[32]。另外,在早发性POAG中也发现了CYP1B1基因突变[33],而 MYOC 基因突变可能与 PCG 相关[34-35]。
1.4 CC CC是一组以晶状体代谢异常为特征的异质性遗传病,临床症状表现为从出生就发生白内障,进行性影响视力甚至致盲。发病率为1/10000~8/10000,其中约1/4与遗传相关。主要是常染色体显性遗传,也有少数是常染色体隐性遗传或X染色体连锁遗传。目前已报道的致病基因至少有29个[36-38]。
2 眼遗传病分子诊断的常用技术
眼遗传病分子诊断最常用的技术是基因全长测序,优点是基因突变的检出率较高,缺点是耗时。如果已知致病基因存在突变热点,则可以先通过一些简单的方法检测,如限制性片段长度多态性(restriction fragment length polymorphism,RFLP),单链构象多态性(single-strand conformational polymorphism,SSCP)[46]等。如果致病基因中存在大片段的缺失或插入,可以用多重连接探针扩增技术(multiplex ligation-dependent probe amplification,MLPA)对DNA序列进行定性和半定量分析[47]。甲基化特异性多重连接探针扩增技术(methylation-specific multiplex ligation-dependent probe amplification,MS-MLPA)还可以对基因启动子区的甲基化程度进行定性和半定量分析[48]。如果DNA检测没有发现突变,则需要提取RNA进行全长测序,检测是否存在内含子剪接突变或大片段重排[49]。通过荧光定量聚合酶链反应(PCR)技术检测RNA水平,可以反映基因表达的变化。
目前,眼遗传病的分子诊断主要局限于DNA检测。然而,随着研究的深入,人们发现蛋白功能、信号通路和基因调节在眼遗传病发病机制中的地位越来越重要,因此,分子诊断的内容和技术也将相应发生改变。
3 眼遗传病分子诊断体系
眼遗传病分子诊断的流程如图1所示。
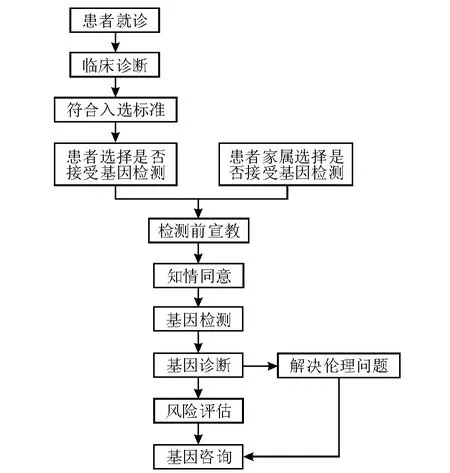
图1.眼遗传病分子诊断流程图
3.1 基因诊断 患者就诊,经临床诊断后,或同家属一起接受基因检测。经过检查前宣教,向患者及其家属解释基因检测的优缺点,解决潜在的伦理问题。受检查者或其合法监护人签署知情同意书。由医护人员抽取受检查者的外周血,至于抗凝管中保存,及时抽提DNA或RNA,进行基因检测。根据检测结果作出基因诊断。
3.2 风险评估 基因检测结果可以用于评估疾病风险。风险评估的意义在于趋利避害,指导患者进行个性化健康管理,采取个性化的行为生活方式,提前做好预防和保健工作,最终达到远离疾病的目的。由于多数常见眼遗传病属于多基因遗传病,而且存在遗传异质性和地域、民族差异,很难明确各种眼遗传病致病基因的突变位点和表型间的关系,这为眼遗传病的风险评估带来了一定的困难。
运用遗传学的基本原则可以预测后代遗传父母突变基因的概率。我们在这里列举最简单的例子,单基因的单点突变。如果父母双方都是同一个基因突变的纯合子,那么后代遗传该突变的概率是100%;如果父母一方是纯合子,另一方是杂合子,其后代有50%的可能是纯合子,同时有50%的可能是杂合子,但不会有正常基因型的后代;如果父母双方都是杂合子,其后代有25%的可能是纯合子,50%的可能是杂合子,还有25%的可能是正常基因型;如果父母一方正常,另一方是杂合子,其后代有50%的可能是杂合子,同时有50%的可能是正常的。
在进行风险评估时还要考虑到疾病的遗传方式,隐性遗传病的杂合子是没有临床症状的携带者,而显性遗传病的杂合子也会出现显著的临床症状。线粒体遗传属于母系遗传,母亲的线粒体基因突变几乎100%遗传给后代,而父亲的线粒体基因突变则不会遗传给后代。
3.3 基因咨询 基因咨询是眼遗传病分子诊断必需的一步。在对患者或其家属进行基因咨询前,需要先得到基因检测结果,并明确基因型和表型的对应关系和疾病的遗传方式。基因咨询可以为准父母提供眼遗传病的基因信息,并帮助他们确定将这些疾病遗传给孩子的可能性。
有些基因突变并不能直接导致疾病的发生,而需要和某些特定的环境因子共同作用诱发疾病。基因咨询不仅能提前告知检测对象患病风险的高低,还能提供个性化健康指导服务、个性化用药指导服务和个性化体检指导服务。帮助人们在疾病发生之前进行准确的预防,通过调整膳食营养、改变生活方式、增加体检频度、接受早期诊治等多种方法,有效避免诱发疾病的环境因素[50]。
4 结语
目前,基因检测还不是医院常规的临床检查项目,分子诊断在眼遗传病的临床应用更是处于起步阶段,但随着对疾病分子基因学研究的深入和高通量分子诊断技术的不断发展,越来越多的眼遗传病的基因信息将被识别并用于指导临床预测、预防、诊断和治疗。因此,急需建立一个标准的分子诊断程序用于眼遗传病的致病基因鉴定。在此之前必须先建立一个种族特异性的眼遗传病数据库,包括临床信息、致病基因及突变位点信息、疾病的遗传方式等,这样才能保证分子诊断更加安全有效地应用于症状前诊断、产前诊断和基因咨询。
[1]MacDonald IM,Tran M,Musarella MA.Ocular genetics:current understanding[J].Surv Ophthalmol,2004,49(2):159-196.
[2]Fan BJ,Tam PO,Choy KW,et al.Molecular diagnostics of genetic eye diseases[J].Clin Biochem,2006,39(3):231-239.
[3]Lohmann DR,Gallie BL.Retinoblastoma:revisiting the model prototype of inherited cancer[J].Am J Med Genet C Semin Med Genet,2004,129C(1):23-28.
[4]Dávalos-Salas M,Furlan-Magaril M,González-Buendía E,et al.Gain of DNA methylation is enhanced in the absence of CTCF at the human retinoblastoma gene promoter[J].BMC Cancer,2011,11:232.
[5]Choy KW,Lee TC,Cheung KF,et al.Clinical implications of promoter hypermethylation in RASSF1A and MGMT in retinoblastoma[J].Neoplasia,2005,7(3):200-206.
[6]Mohney BG,Robertson DM,Schomberg PJ,et al.Second nonocular tumors in survivors of heritable retinoblastoma and prior radiation therapy[J].Am J Ophthalmol,1998,126(2):269-277.
[7]Rivolta C,Sharon D,DeAngelis MM,et al.Retinitis pigmentosa and allied diseases:numerous diseases,genes,and inheritance patterns[J].Hum Mol Genet,2002,11(10):1219-1227.
[8]Wang DY,Chan WM,Tam PO,et al.Gene mutations in retinitis pigmentosa and their clinical implications[J].Clin Chim Acta,2005,351(1/2):5-16.
[9]Kajiwara K,Berson EL,Dryja TP.Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci[J].Science,1994,264(5165):1604-1608.
[10]Katsanis N,Ansley SJ,Badano JL,et al.Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder[J].Science,2001,293(5538):2256-2259.
[11]Mansergh FC,Millington-Ward S,Kennan A,et al.Retinitis pigmentosa and progressive sensorineural hearing loss caused by a C12258A mutation in the mitochondrial MTTS2 gene[J].Am J Hum Genet,1999,64(4):971-985.
[12]Sohocki MM,Daiger SP,Bowne SJ,et al.Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies[J].Hum Mutat,2001,17(1):42-51.
[13]Berson EL,Grimsby JL,Adams SM,et al.Clinical features and mutations in patients with dominant retinitis pigmentosa-1(RP1)[J].Invest Ophthalmol Vis Sci,2001,42(10):2217-2224.
[14]Musarella MA,Anson-Cartwright L,Leal SM,et al.Multipoint linkage analysis and heterogeneity testing in 20 X-linked retinitis pigmentosa families[J].Genomics,1990,8(2):286-296.
[15]Young TK,Souzeau E,Liu L,et al.Compound heterozygote myocilin mutations in a pedigree with high prevalence of primary open-angle glaucoma[J].Mol Vis,2012,18:3064-3069.
[16]Izzotti A,Di Marco B,De Flora S,et al.Open angle glaucoma:epidemiology,pathogenesis and prevention[J].Recenti Prog Med,2006,97(1):37-45.
[17]Harris D.The inheritance of glaucoma.A pedigree of familial glaucoma[J].Am J Ophthalmol,1965,60:91-95.
[18]Sarfarazi M,Stoilov I.Molecular genetics of primary congenital glaucoma[J].Eye(Lond),2000,14(Pt 3B):422-428.
[19]Stone EM,Fingert JH,Alward WL,et al.Identification of a gene that causes primary open angle glaucoma[J].Science,1997,275(5300):668-670.
[20]Rezaie T,Child A,Hitchings R et al.Adult-onset primary openangle glaucoma caused by mutations in optineurin[J].Science,2002,295(5557):1077-1079.
[21]Monemi S,Spaeth G,DaSilva A,et al.Identification of a novel adult-onset primary open-angle glaucoma(POAG)gene on 5q22.1[J].Hum Mol Genet,2005,14(6):725-733.
[22]Stoilov I,Akarsu AN,Sarfarazi M.Identification of three different truncating mutations in cytochrome P4501B1(CYP1B1)as the principal cause of primary congenital glaucoma(Buphthalmos)in families linked to the GLC3A locus on chromosome 2p21.Hum Mol Genet,1997,6(4):641-647.
[23]Narooie-Nejad M,Paylakhi SH,Shojaee S,et al.Loss of function mutations in the gene encoding latent transforming growth factor beta binding protein 2,LTBP2,cause primary congenital glaucoma[J].Hum Mol Genet,2009,18(20):3969-3977.
[24]Ali M,McKibbin M,Booth A,et al.Null mutations in LTBP2 cause primary congenital glaucoma[J].Am J Hum Genet,2009,84(5):664-671.
[25]Cheng JW,Cheng SW,Ma XY,et al.,Myocilin polymorphisms and primary open-angle glaucoma:a systematic review and meta-analysis[J].PLoS One,2012,7(9):e46632.
[26]Orwig SD,Perry CW,Kim LY,et al.Amyloid fibril formation by the glaucoma-associated olfactomedin domain of myocilin[J].J Mol Biol,2012,421(2/3):242-255.
[27]Fan BJ,Wang DY,Fan DS,et al.SNPs and interaction analyses of myocilin,optineurin,and apolipoprotein E in primary open angle glaucoma patients[J].Mol Vis,2005,11:625-631.
[28]Chen JH,Xu L,Li Y.Study on the optic neuropathy induced response protein gene mutation in Chinese patients with primary open-angle glaucoma[J].Zhonghua Yi Xue Za Zhi,2004,84(13):1098-1102.
[29]Leung YF,Fan BJ,Lam DS,et al.Different optineurin mutation pattern in primary open-angle glaucoma[J].Invest Ophthalmol Vis Sci,2003,44(9):3880-3884.
[30]Fan BJ,Wang DY,Cheng CY,et al.Different WDR36 mutation pattern in Chinese patients with primary open-angle glaucoma[J].Mol Vis,2009,15:646-653.
[31]Colomb E,Kaplan J,Garchon HJ.Novel cytochrome P4501B1(CYP1B1)mutations in patients with primary congenital glaucoma in France[J].Hum Mutat,2003,22(6):496.
[32]Kakiuchi-Matsumoto T,Isashiki Y,Ohba N,et al.Cytochrome P4501B1 gene mutations in Japanese patients with primary congenital glaucoma(1)[J].Am J Ophthalmol,2001,131(3):345-350.
[33]Vincent AL,Billingsley G,Buys Y,et al.Digenic inheritance of early-onset glaucoma:CYP1B1,a potential modifier gene[J].Am J Hum Genet,2002,70(2):448-460.
[34]Kaur K,Reddy AB,Mukhopadhyay A,et al.Myocilin gene implicated in primary congenital glaucoma[J].Clin Genet,2005,67(4):335-340.
[35]Kennedy KD,AnithaChristy SA,Buie LK,et al.Cystatin a,a potential common link for mutant myocilin causative glaucoma[J].PLoS One,2012,7(5):e36301.
[36]Litt M,Carrero-Valenzuela R,LaMorticella DM,et al.Autosomal dominant cerulean cataract is associated with a chain termination mutation in the human beta-crystallin gene CRYBB2[J].Hum Mol Genet,1997,6(5):665-668.
[37]Litt M,Kramer P,LaMorticella DM,et al.Autosomal dominant congenital cataract associated with a missense mutation in the human alpha crystallin gene CRYAA[J].Hum Mol Genet,1998,7(3):471-474.
[38]Qi Y,Jia H,Huang S,et al.A deletion mutation in the betaA1/A3 crystallin gene(CRYBA1/A3)is associated with autosomal dominant congenital nuclear cataract in a Chinese family[J].Hum Genet,2004,114(2):192-197.
[39]Pang CP,Leung YF,Fan B,et al.TIGR/MYOC gene sequence alterations in individuals with and without primary open-angle glaucoma[J].Invest Ophthalmol Vis Sci,2002,43(10):3231-3235.
[40]Gong G,Kosoko-Lasaki O,Haynatzki GR,et al.Genetic dissection of myocilin glaucoma[J].Hum Mol Genet,2004,13 Spec No 1:R91-102.
[41]Fan BJ,Leung DY,Wang DY,et al.Novel myocilin mutation in a Chinese family with juvenile-onset open-angle glaucoma[J].Arch Ophthalmol,2006,124(1):102-106.
[42]Bejjani BA,Lewis RA,Tomey KF,et al.Mutations in CYP1B1,the gene for cytochrome P4501B1,are the predominant cause of primary congenital glaucoma in Saudi Arabia[J].Am J Hum Genet,1998,62(2):325-333.
[43]Sitorus R,Ardjo SM,Lorenz B,et al.CYP1B1 gene analysis in primary congenital glaucoma in Indonesian and European patients[J].J Med Genet,2003,40(1):e9.
[44]Kumar A,Duvvari MR,Prabhakaran VC,et al.A homozygous mutation in LTBP2 causes isolated microspherophakia[J].Hum Genet,2010,128(4):365-371.
[45]Lohmann DR.RB1 gene mutations in retinoblastoma[J].Hum Mutat,1999,14(4):283-288.
[46]Dong Y,Zhu H.Single-strand conformational polymorphism analysis:basic principles and routine practice[J].Methods Mol Med,2005,108:149-157.
[47]Hömig-Hölzel C,Savola S.Multiplex ligation-dependent probe amplification(MLPA)in tumor diagnostics and prognostics[J].Diagn Mol Pathol,2012,21(4):189-206.
[48]Nygren AO,Ameziane N,Duarte HM,et al.Methylation-specific MLPA(MS-MLPA):simultaneous detection of CpG methylation and copy number changes of up to 40 sequences[J].Nucleic Acids Res,2005,33(14):e128.
[49]Vreeswijk MP,van der Klift HM.Analysis and interpretation of RNA splicing alterations in genes involved in genetic disorders[J].Methods Mol Biol,2012,867:49-63.
[50]Ross LF,Saal HM,David KL,et al.Technical report:Ethical and policy issues in genetic testing and screening of children[J].Genet Med,2013,15(3):234-245.
猜你喜欢
中国生殖健康(2020年2期)2021-01-18
中学生理科应试(2019年4期)2019-07-08
科学之谜(2019年3期)2019-03-28
中国生殖健康(2019年3期)2019-02-01
科学之谜(2018年8期)2018-09-29
小学生导刊(2018年13期)2018-06-29
中国生殖健康(2018年2期)2018-01-12
恋爱婚姻家庭·养生版(2016年9期)2016-09-07
医学研究杂志(2015年12期)2015-06-10
中央民族大学学报(自然科学版)(2015年2期)2015-06-09
