
儿童2型Dent病1例并文献复习
2014-08-10 12:28李国民方晓燕茜孙利翟亦晖郭慕依吴冰冰
中国循证儿科杂志 2014年6期
李国民 方晓燕 徐 虹 沈 茜孙 利翟亦晖郭慕依 安 宇 吴冰冰
·论著·
儿童2型Dent病1例并文献复习
李国民1,4方晓燕1,4徐 虹1沈 茜1孙 利1翟亦晖1郭慕依2安 宇3吴冰冰3
目的 总结1例2型Dent病患儿临床资料,提高对该病的认识。方法 报道1例2型Dent病患儿的临床发现、相关实验室检查指标和肾活检病理改变。对该家系相关成员行CLCN5和OCRL基因外显子及附近调控区域直接测序,分析突变位点,并文献复习。结果 患儿,男,6岁起病,首发症状为单纯性蛋白尿,未见先天性白内障、智力低下、认知障碍及发育迟缓。实验室检查提示低分子蛋白尿,高钙尿症,镜下血尿,血清乳酸脱氢酶和磷酸肌酸激酶增高,肾活检病理提示轻微病变。患儿CLCN5和OCRL基因测序分析发现,CLCN5基因未见致病性突变,ORCL基因c.260delA(p.Q87fs105X)纯合突变,确诊为2型Dent病。家系OCRL基因突变分析显示,患儿父亲c.260delA未检出突变,母亲携带c.260delA杂合突变。c.260delA突变为移码突变,可导致OCRL基因编码蛋白截短。结论 2型Dent病以低分子蛋白尿、高钙尿症和镜下血尿为特征,血清乳酸脱氢酶、磷酸肌酸激酶增高和OCRL基因纯合移码突变支持2型Dent病诊断。
2型Dent病;CLCN5基因;OCRL基因
1 病例资料
男,汉族,2004年6月出生,2010年10月因“体检发现蛋白尿3个月”就诊复旦大学附属儿科医院(我院),以蛋白尿待查收入我院。
患儿3个月前体检时发现尿蛋白2+,不伴有发热、腹痛,无尿频、尿急、尿痛,未见水肿及少尿,未见肉眼血尿。当地医院多次复查尿常规,尿蛋白均阳性(+~3+),24 h尿蛋白定量在1.40~1.45 g·L-1,予贝那普利、黄芪颗粒治疗,尿蛋白未转阴。
患儿系G1P1,足月剖宫产,出生无产伤及窒息,出生体重4 100 g。既往体健,无肾脏病病史。父母均体健,非近亲结婚,家族成员中无类似疾病史。
入院查体:身高120 cm,体重21 kg。血压90/60 mmHg,神志清楚,精神尚可,全身未见水肿,两肺及心脏听诊正常,腹平坦,肾区无叩击痛,肋下未扪及肝、脾肿大。神经系统检查无认知障碍,智力测试正常,眼科检查未发现角膜色素沉着和白内障。
血电解质、肝肾功能、血胆固醇正常。血清乳酸脱氢酶278 U·L-1,磷酸肌酸激酶42 U·L-1。尿常规蛋白2+,镜下血尿,24 h尿蛋白定量1.02 g·L-1,α1微球蛋白79 mg·L-1。24 h尿Ca 5.31 mg·kg-1,尿Ca/Cr 0.43。泌尿系统B超示肾脏结构和大小未见异常,未见结石和钙盐沉着。肾活检示:光镜下可见30个肾小球,除1个肾小球硬化外,余肾小球形态结构大致正常,肾小管无异常(图1A,B)。免疫荧光提示IgG、IgA、IgM、C3、C4、C1q和Fb均阴性,Ⅳ型胶原α1、α3和α5链均阳性。电镜下可见5个肾小球,未有其他异常发现(图1C)。

图1 本文患儿肾脏病理改变
Fig 1 Renal pathological changes in the proband
Notes Globally sclerosed glomerulus shown in A (HE stain original magnification 200×), minute lesion shown in B ( PAS stain original magnification 200×) under light microscope; Normal glomerular structure shown in C under electron microscopy ( original magnification 11 500×)
对患儿(先证者)家系采用“2次试纸法”行尿液筛查。该家系共有3代成员26人,本例患儿尿液筛查尿蛋白阳性,其母亲尿蛋白可疑阳性(±),余24位家系成员2次尿液筛查均阴性。利用Sanger法首先对先证者及其父母行CLCN5基因外显子和其附近区域进行直接测序,未发现致病性突变。 对OCRL基因外显子和其附近区域进行直接测序,测序结果与美国国家生物技术信息中心(NCBI)GenBank (http://www.ncbi.nlm.nih.gov/Genbank/) 中的序列进行比对,其中核苷酸序列参照 NG_008638.1OCRL,cDNA序列参照 NM_0000276.3OCRL。氨基酸序列参照Uni-ProtKB (http://www.uniprot.org/uniprot/Q01968, ID: Q01968) 和 the Ensembl Genome Browser (http://www.ensembl.org, ID: ENSP00000360154) 提供的序列进行比对。测序结果显示(图2),患儿OCRL基因5号外显子c.260delA (p.Q87fs105X)纯合缺失变异,确诊为2型Dent病,患儿母亲携带杂合c.260delA变异,其父亲未发现c.260delA变异。在100例正常人群中未发现有c.260delA变异。c.260delA变异是单个碱基的缺失,引起移码突变,造成OCRL基因编码蛋白截短。其余家系成员尿蛋白阴性未行基因突变分析。

图2 患儿及其父母OCRL基因检测结果
Fig 2 Renal pathological changes in proband
Notes Mutational analysis ofOCRLin the family. Arrows indicate mutation. A: control, B: proband, C: mother, D: father
予患儿依那普利、氢氯噻嗪口服,并嘱多饮水,并定期复查肾功能。患儿出院后3和4年来我院随访,血电解质和血气分析正常;血清乳酸脱氢酶300~324 U·L-1,磷酸肌酸激酶32~302 U·L-1;仍有镜下血尿,24 h尿蛋白定量1.31~1.54 g·L-1,α1微球蛋白275~528.3 mg·L-1,24 h尿Ca 5.31~9.13 mg·kg-1,尿Ca/Cr 0.41~0.44;肾脏B超未见结石和钙盐沉积。随访患儿母亲24 h尿蛋白定量0.18 g·L-1,磷酸肌酸激酶202 U·L-1,乳酸脱氢酶265 U·L-1;肝、肾功能、电解质和血气分析均正常。
2 讨论
Dent病是一种罕见的X连锁隐性遗传性肾小管疾病,以近端小管功能异常为表现,其主要特征为低分子量蛋白尿(LMWP)、高钙尿症、肾结石、肾钙质沉积症和肾功能异常[1, 2]。尽管Dent病累及近端肾小管功能,但极少患者有糖尿、氨基酸尿、高磷酸盐尿和尿液酸化功能障碍等范可尼综合征的表现[3]。典型Dent病症状仅见于男性,一般在儿童早期就可出现[4]。<10岁的儿童可表现为LMWP和(或)镜下血尿,症状隐匿。30%~80%的Dent病患者于30~50岁进展为终末期肾病(ESRD),然而也有部分患者至60岁以上也未进展为ESRD[1, 4, 5]。女性携带者症状轻微,可仅有镜下血尿,罕有进展为ESRD的报道[1, 6]。佝偻病或软骨病是Dent病常见的并发症。典型Dent病的诊断需同时符合Hoopes标准[7]中的3条:①LMWP:尿中低分子蛋白至少升高5倍,用于监测的低分子蛋白主要有视黄醇结合蛋白、α1微球蛋白和β2微球蛋白;②高钙尿症:24 h尿Ca定量>4.0 mg·kg-1或随机尿Ca/Cr>0.25 mg·mg-1;③有下列情况之一:镜下血尿、肾结石、肾钙盐沉着症、低磷血症和肾功能异常。本文患儿临床特征为LMWP,α1微球蛋白定量异常增高;高钙尿症,24 h尿Ca定量和尿Ca/Cr均高于同年龄儿童的正常值;持续性镜下血尿。因此,本文患儿符合Dent病的临床诊断。
约65%的Dent病由CLCN5基因突变引起(1型Dent病, OMIM #300009)[1, 2]。另有15%的Dent病由OCRL基因突变引起(2型Dent病, MIM #300555)[2, 8, 9],余下的20% Dent病尚未发现致病基因[5, 8]。Dent病发病率目前尚不清楚,以“Dent disease”为检索词检索PubMed数据库,1型Dent病病例约250例,2型Dent病有57例病例报道[2]。国内曾有1型Dent病的报道[6, 10, 11],尚未见2型Dent病报道。
CLCN5基因位于染色体Xp11.22-11.23,编码746个氨基酸长度的CLC-5蛋白[12]。CLC-5蛋白属于电压门控性氯离子通道蛋白家族成员,具有高度保守的结构,在体内发挥着重要作用,包括调节膜的兴奋性、维持细胞内外离子的稳态和调节酸化功能[13~16]。OCRL基因位于染色体Xq25,编码磷脂酰肌醇4,5-二磷酸5-磷酸酶。该酶可以水解磷脂酰肌醇4,5-二磷酸(PIP2)[7, 17]。参与调节肌动蛋白聚合过程,进而影响细胞迁移和细胞间接触,在肾小管、眼晶状体和脑组织等发育过程中起重要作用[2, 18]。本文患儿CLCN5基因突变分析未发现致病性突变,OCRL基因突变分析发现5号外显子c.260delA (p.Q87fs105X)纯合缺失变异。在100例正常人群中未发现有c.260delA变异。c.260delA变异是单个碱基的缺失,引起移码突变,造成OCRL基因编码蛋白截短,为致病性突变。故患儿发病与致病性c.260delA纯合突变有关。人类基因突变资料库未发现该突变报道(http://www.hgmd.cf.ac.uk/), 应为新的突变。家系OCRL基因突变分析提示,患儿母亲有c.260delA杂合突变,为携带者,除α1微球蛋白、24 h尿蛋白定量、磷酸肌酸激酶和乳酸脱氢酶偏高外,无其他临床表现。
OCRL基因最早被发现是Lowe 综合征(眼-脑-肾综合征, OMIM #309000)的致病基因[18]。Lowe 综合征典型的临床特征为双侧先天性白内障、近端小管功能异常(常表现为范可尼综合征)、精神和运动发育落后[19, 20]。PubMed数据库检索到的57例2型Dent病[5,7,17,21~29]和本文报道的1例汇总分析结果如表1所示,18/58例2型Dent病患者有轻度智力障碍,发育延迟等肾外症状,尚未见严重的精神和运动发育落后报道,58例均无肾小管酸中毒、糖尿、氨基酸等范可尼综合征的表现,4例有外周性白内障,但无先天性白内障。27/54例伴血清磷酸肌酸激酶和乳酸脱氢酶的增高。本文患儿无眼部和脑部异常发现,近端肾小管障碍表现为LMWP和高钙尿症,未见糖尿、氨基酸尿等范可尼综合征的表现,血气分析提示也无肾小管酸中毒表现,提示符合2型Dent病临床特点。多次血清磷酸肌酸激酶和乳酸脱氢酶均升高,提示肌肉受累。引起2型Dent病的突变位点通常发生在OCRL基因5′端,位于外显子1~7,为磷脂酰肌醇4,5-二磷酸5-磷酸酶结构域编码区,而Lowe 综合征突变位点一般位于OCRL基因的外显子8~24。本文患儿OCRL基因突变位点位于外显子5上,符合2型Dent病遗传学特点[18, 30]。
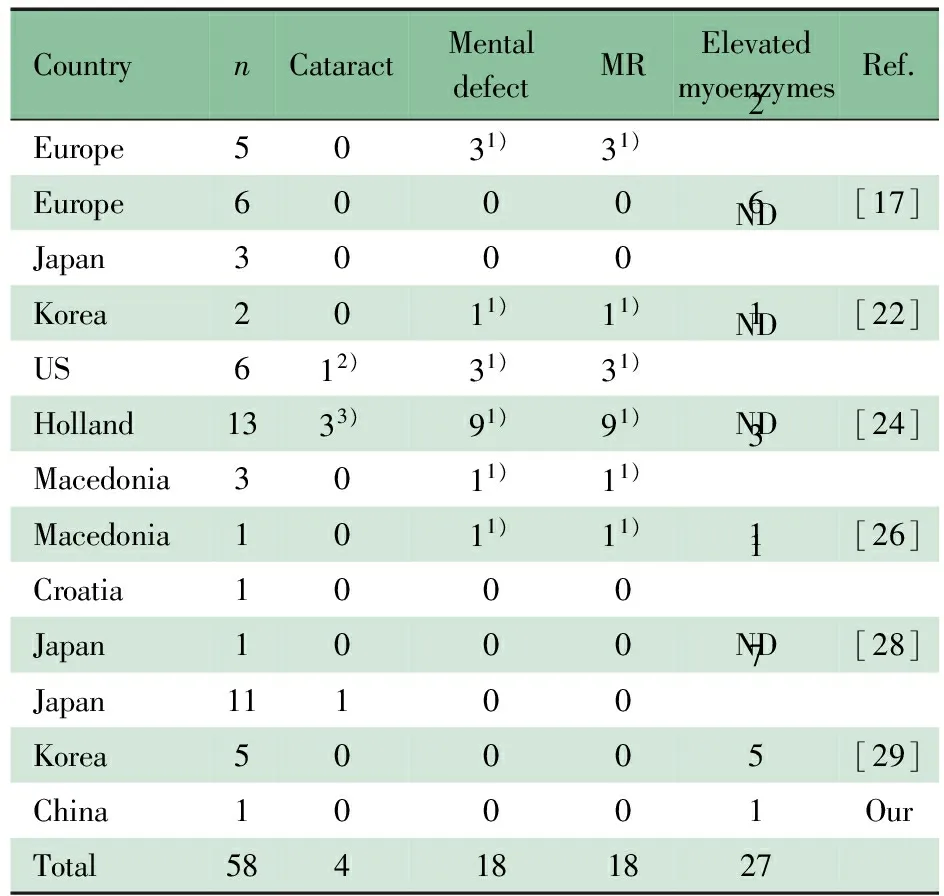
表1 已报道的2型Dent病患者的肾外表现(n)
Notes MR: Mental retardation; ND:not done. 1) mild; 2) normal vision; 3) peripheral early cataract detected by slit-lamp
1型Dent病患者通常表现为LMWP,24 h尿蛋白定量为轻度或中度蛋白尿。近年来有研究发现,少部分低龄1型Dent病可有肾病水平蛋白尿,但这部分患儿无低蛋白血症,尿蛋白电泳表现,LMWP和白蛋白均增高[9]。2型Dent病患者不仅有肾脏表现,还可有肾外表现。1型和2型Dent病的症状相似,主要靠CLCN5和OCRL基因突变分析来鉴别。2型Dent病患者均有磷酸肌酸激酶和乳酸脱氢酶升高,部分患者还伴肾外症状。因此,临床上对肌酶升高或有肾外症状的病例可先行OCRL基因突变分析,而对无肾外症状或肌酶升高的病例应先行CLCN5突变分析。
由于局灶节段肾小球硬化肾病综合征(FSGS-NS)患儿的近端肾小管功能异常比微小病变肾病综合征(MCD-NS)更常见。 据此, 有学者推断FSGS可继发于原发性近端肾小管功能异常或损害[31]。Kaneko等[28]研究也发现,2型Dent病可引起FSGS。本文患儿肾活检结果显示,电镜下5个肾小球结构无明显异常,免疫荧光正常,光镜下30个肾小球,仅1个肾小球硬化,其他肾小球结构大致正常,肾小管无异常改变。1个硬化肾小球周围的肾小球结构大致正常,该硬化肾小球较孤立,推测其是生理状态下的自然改变,与疾病关系不大。本文患儿肾活检并未发现FSGS改变,可能与患儿病程较短有关,不排除进展为FSGS的可能。尽管Dent病患者肾脏病理光镜下有FSGS改变,但电镜下仅表现足突部分融合,而不是广泛融合[9]。因此,FSGS可能不是Dent病特征性的病理改变,而仅仅可能是疾病病程的反映。
[1]Claverie-Martin F, Ramos-Trujillo E, Garcia-Nieto V. Dent's disease: clinical features and molecular basis. Pediatr Nephrol,2011,26(5):693-704
[2]Ludwig M, Levtchenko E, Bokenkamp A. Clinical utility gene card for: Dent disease (Dent-1 and Dent-2). Eur J Hum Genet,2014
[3]Hodgin JB, Corey HE, Kaplan BS, et al. Dent disease presenting as partial Fanconi syndrome and hypercalciuria. Kidney Int,2008,73(11):1320-1323
[4]Devuyst O, Thakker RV. Dent's disease. Orphanet J Rare Dis,2010,5:28
[5]Sekine T, Komoda F, Miura K, et al. Japanese Dent disease has a wider clinical spectrum than Dent disease in Europe/USA: genetic and clinical studies of 86 unrelated patients with low-molecular-weight proteinuria. Nephrol Dial Transplant,2014,29(2):376-384
[6]Zhang H, Wang C, Yue H, et al. Identification of a novel mutation in the CLCN5 gene in a Chinese family with Dent-1 disease. Nephrology (Carlton),2014,19(2):80-83
[7]Hoopes RJ, Shrimpton AE, Knohl SJ, et al. Dent disease with mutations in OCRL1. Am J Hum Genet,2005,76(2):260-267
[8]Grand T, L'Hoste S, Mordasini D, et al. Heterogeneity in the processing of CLCN5 mutants related to Dent disease. Hum Mutat,2011,32(4):476-483
[9]Cramer MT, Charlton JR, Fogo AB, et al. Expanding the phenotype of proteinuria in Dent disease. A case series. Pediatr Nephrol,2014,29(10):2051-2054
[10]Zhu BQ(朱碧溱), Li P, Huang JP. Clinical and genetic analysis of Dent' s disease in 6 Chinese children with low molecular weight proteinuria. Chin J Pediatr(中华儿科杂志),2010,48(5):329-333
[11]Ji L, Chen C, Wang J, et al. A novel CLCN5 mutation in a Chinese boy with Dent's disease. World J Pediatr,2014,10(3):275-277
[12]Lloyd SE, Pearce SH, Fisher SE, et al. A common molecular basis for three inherited kidney stone diseases. Nature,1996,379(6564):445-449
[13]Hoopes RJ, Hueber PA, Reid RJ, et al. CLCN5 chloride-channel mutations in six new North American families with X-linked nephrolithiasis. Kidney Int,1998,54(3):698-705
[14]Picollo A, Pusch M. Chloride/proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature,2005,436(7049):420-423
[15]Lippiat JD, Smith AJ. The CLC-5 2Cl(-)/H(+) exchange transporter in endosomal function and Dent's disease. Front Physiol,2012,3:449
[16]Tosetto E, Casarin A, Salviati L, et al. Complexity of the 5'UTR region of the CLCN5 gene: eleven 5'UTR ends are differentially expressed in the human kidney. BMC Med Genomics,2014,7:41
[17]Utsch B, Bokenkamp A, Benz MR, et al. Novel OCRL1 mutations in patients with the phenotype of Dent disease. Am J Kidney Dis,2006,48(6):941-942
[18]Bokenkamp A, Levtchenko E, Recker F, et al. Clinical utility gene card for: Lowe syndrome. Eur J Hum Genet,2014
[19]Zhang YQ, Wang F, Ding J, et al. Novel OCRL mutations in Chinese children with Lowe syndrome. World J Pediatr,2013,9(1):53-57
[20]Kim HK, Kim JH, Kim YM, et al. Lowe syndrome: a single center's experience in Korea. Korean J Pediatr,2014,57(3):140-148
[21]Sekine T, Nozu K, Iyengar R, et al. OCRL1 mutations in patients with Dent disease phenotype in Japan. Pediatr Nephrol,2007,22(7):975-980
[22]Cho HY, Lee BH, Choi HJ, et al. Renal manifestations of Dent disease and Lowe syndrome. Pediatr Nephrol,2008,23(2):243-249
[23]Shrimpton AE, Hoopes RJ, Knohl SJ, et al. OCRL1 mutations in Dent 2 patients suggest a mechanism for phenotypic variability. Nephron Physiol,2009,112(2):27-36
[24]Bokenkamp A, Bockenhauer D, Cheong HI, et al. Dent-2 disease: a mild variant of Lowe syndrome. J Pediatr,2009,155(1):94-99
[25]Tasic V, Lozanovski VJ, Korneti P, et al. Clinical and laboratory features of Macedonian children with OCRL mutations. Pediatr Nephrol,2011,26(4):557-562
[26]Lozanovski VJ, Ristoska-Bojkovska N, Korneti P, et al. OCRL1 mutation in a boy with Dent disease, mild mental retardation, but without cataracts. World J Pediatr,2011,7(3):280-283
[27]Vrljicak K, Batinic D, Milosevic D, et al. A boy with Dent-2 disease. Coll Antropol,2011,35(3):925-928
[28]Kaneko K, Hasui M, Hata A, et al. Focal segmental glomerulosclerosis in a boy with Dent-2 disease. Pediatr Nephrol,2010,25(4):781-782
[29]Park E, Choi HJ, Lee JM, et al. Muscle involvement in Dent disease 2. Pediatr Nephrol,2014,29(11):2127-2132
[30]Hichri H, Rendu J, Monnier N, et al. From Lowe syndrome to Dent disease: correlations between mutations of the OCRL1 gene and clinical and biochemical phenotypes. Hum Mutat,2011,32(4):379-388
[31]Copelovitch L, Nash MA, Kaplan BS. Hypothesis: Dent disease is an underrecognized cause of focal glomerulosclerosis. Clin J Am Soc Nephrol,2007,2(5):914-918
(本文编辑:丁俊杰)
Dent-2 disease in one child and literature review
LIGuo-min1,4,FANGXiao-yan1,4,XUHong1,SHENQian1,SUNLi1,ZHAIYi-hui1,GUOMu-yi2,ANYu3,WUBing-bing3
(1DepartmentofKidneyandRheumatology,Children'sHospitalofFudanUniversity,Shanghai201102; 2DepartmentofPathology,ShanghaiMedicalCollegeofFudanUniversity,Shanghai200023; 3MedicalTranslationalCenterofChildren'sHospitalofFudanUniversity,Shanghai201102,China; 4hasequalcontribution)
Corresponding Author:XU Hong,E-mail:hxu@shmu.edu.cn
ObjectiveTo summarize and review the clinical data of a child with Dent-2 disease so as to improve our understanding to the disease.Methods Clinical data of the case with Dent-2 disease were summarized, including clinical manifestations, laboratory findings, renal pathological changes and family investigation. Mutation analysis inCLCN5 andOCRLgenes was performed by direct sequencing in his family. Mutations ofCLCN5 andOCRLgenes were examined in normal control and related literatures from PubMed were reviewed also.ResultsAge at onset was 6 years old. Proteinuria was the only symptom at onset. The patient had no extrarenal symptoms of Lowe syndrome, such as peripheral cataracts, mental impairment and stunted growth. Laboratory findings showed low-molecular weight proteinuria, hypercalciuria, microscopic hematuria and elevation of creatine kinase/lactate dehydrogenase. Renal pathological change detection showed a minute lesion under light microscope. A novel homozygous c.260delA (p.Q87fs105X) mutation was found in exon 5 ofOCRLgene. No mutations ofCLCN5 gene were detected. Mutation analysis showed his mother carried a c.260delA mutation, but his father did not. This deletion caused a frameshift of amino acids and resulted in a truncated protein (p.Q87fs105X), and was not identified in the 100 healthy, unrelated controls.ConclusionDent-2 disease is an X-linked tubulopathy characterized by low-molecular-weight proteinuria (LMWP), hypercalciuria, nephrocalcinosis, nephrolithiasis, and the development of renal insufficiency. Dent-2 disease can be diagnosed by these characteristics. Lowe syndrome was ruled out in the patient with no peripheral cataracts, mental impairment and stunted growth. Elevation of creatine kinase/lactate dehydrogenase and carrying the homozygous frameshift mutation inOCRLgene support the diagnosis of the patient in the current study. This is the first report of Dent-2 disease caused byOCRLgene mutation in Chinese family.
Dent-2 disease;CLCN5 gene;OCRLgene
复旦大学附属儿科医院人才工程-学科带头人(1125)培育计划项目
1 复旦大学附属儿科医院肾脏风湿科 上海,201102; 2 复旦大学上海医学院病理教研室 上海,200032;3 复旦大学附属儿科医院医学转化中心 上海,201102;4 共同第一作者
徐虹,E-mail:hxu@shmu.edu.cn
10.3969/j.issn.1673-5501.2014.06.011
2014-10-03
2014-11-20)
猜你喜欢
祝您健康·文摘版(2021年12期)2021-12-08
冰雪运动(2021年1期)2021-07-28
家庭科学·新健康(2021年5期)2021-06-21
科学大众(2021年10期)2021-05-20
岭南急诊医学杂志(2020年6期)2021-01-14
安徽医专学报(2020年3期)2020-12-25
医学信息(2016年29期)2016-11-28
中国医药生物技术(2015年4期)2015-12-26
当代体育科技(2015年8期)2015-02-27
祝您健康(2000年11期)2000-12-31
