
HPLC法测定盐酸替罗非班原料药的含量及有关物质
2015-03-09 10:37张福成解放军空军总医院北京100142
中国药房 2015年3期
吴 燕,周 敏,张福成(解放军空军总医院,北京 100142)
抗血小板治疗在冠状动脉粥样硬化性心脏病治疗中起着十分重要的作用。盐酸替罗非班是一种非肽类的血小板糖蛋白Ⅱb/Ⅲa受体(该受体是与血小板聚集过程有关的主要血小板表面受体)的可逆性拮抗药,可阻止纤维蛋白原与糖蛋白Ⅱb/Ⅲa受体结合,从而阻断血小板的交联及血小板的聚集[1-3]。盐酸替罗非班可抑制二磷酸腺甘诱导的血小板聚集及延长出血时间,可强效抑制血小板功能,作用显著,特异性强。目前,关于盐酸替罗非班原料药和注射剂的质量标准未见文献报道。为此,本研究建立了以高效液相色谱(HPLC)法测定盐酸替罗非班原料药的含量及有关物质的方法。
1 材料
LC-10AD HPLC仪,包括SPD-10AVP检测器、CLASSLC 10A色谱工作站(日本岛津公司);AE-240万分之一电子天平(瑞士梅特勒-托利多公司)。
盐酸替罗非班对照品(中国食品药品检定研究院,批号:100646-200401,纯度:99.9%);盐酸替罗非班原料药(解放军空军总医院自制,批号:201230601、201230602、201230603,质量分数:99.5%、99.6%、99.7%);盐酸替罗非班中间体Ⅰ、Ⅱ、Ⅲ(解放军空军总医院自制,批号均为:201230601,纯度分别为:99.4%、99.5%、99.4%);甲醇、乙腈为色谱纯,其他试剂为分析纯,水为屈臣氏水。
2 方法与结果
2.1 色谱条件
色谱柱:CAPCELL PAK C18(250 mm×4.6 mm,5 μm);流动相:流动相A为0.1%醋酸铵溶液(pH 2.30)-乙腈-甲醇(90 ∶5∶5,V/V/V),流动相B为0.1%醋酸铵溶液(pH 2.30)-乙腈-甲醇(10 ∶85 ∶5,V/V/V),梯度洗脱程序见表1;检测波长:227 nm;柱温:35 ℃;流速:1.0 ml/min;进样量:10µl。
2.2 含量测定方法
取样品(批号:201230601)适量,用流动相B定量稀释制成0.1 mg/ml的溶液,作为供试品溶液,精密量取10µl进样测定;另取盐酸替罗非班对照品适量,同法制备对照品溶液,进样测定,记录色谱,详见图1。按外标法以峰面积计算含量。

表1 梯度洗脱程序Tab 1 Gradient elution program
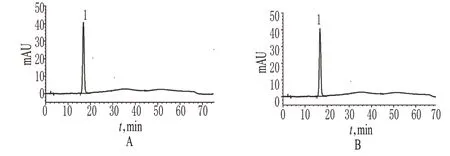
图1 高效液相色谱图A.对照品溶液;B.供试品溶液;1.盐酸替罗非班Fig 1 HPLC chromatogramsA.substance control;B.test sample;1.tirofiban hydrochloride
2.3 有关物质测定方法
取样品(批号:201230601)适量,加流动相B稀释制成1 mg/ml的溶液,作为供试品溶液;精密量取1 ml,置于100 ml量瓶中,加流动相B稀释至刻度,摇匀,作为对照溶液。取对照溶液10 μl注入HPLC仪,调节检测灵敏度,使主成分色谱峰高约为满量程的20%;再精密量取供试品溶液与对照溶液各10 μl,分别注入HPLC仪,记录色谱,详见图2。供试品溶液的色谱图中如有杂质峰,按自身对照法计算有关物质的量。
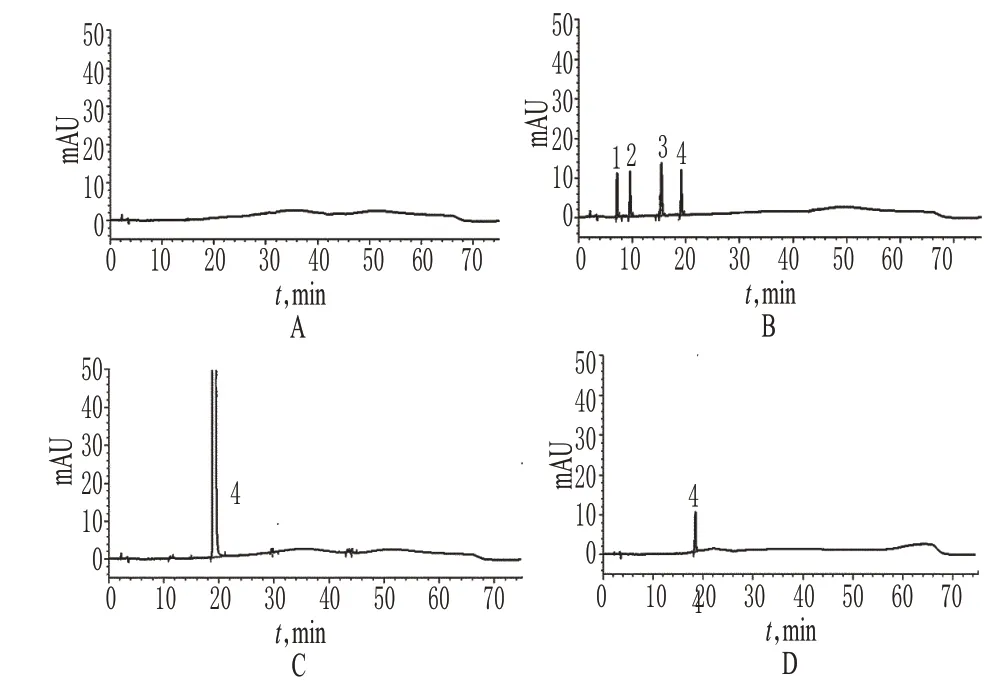
图2 有关物质测定和系统适用性试验高效液相色谱图A.空白溶剂(水);B.系统适用性溶液;C.供试品溶液;D.对照溶液;1.中间体Ⅰ;2.中间体Ⅲ;3.中间体Ⅱ;4.盐酸替罗非班Fig 2 HPLC chromatograms of related substance and system suitabilityA.blank solvent(water);B.system suitability solution;C.test sample;D.substance control;1.intermediateⅠ;2.intermediate Ⅲ;3.intermediateⅡ;4.tirofiban hydrochloride
2.4 系统适用性试验
分别称取盐酸替罗非班中间体Ⅰ、Ⅱ、Ⅲ各10 mg,置于10 ml量瓶中,用流动相B定容,得系统适用性贮备液;精密称取样品(批号:201230601)100 mg,置于100 ml量瓶中,精密移取系统适用性贮备液1 ml置于此量瓶中,定容,摇匀,得系统适用性溶液。量取系统适用性溶液10 μl注入HPLC仪,记录色谱,详见图2。结果表明,在“2.1”项色谱条件下,盐酸替罗非班与各中间体及各个中间体之间的分离度均>1.5,分离度良好。
2.5 方法专属性考察
取样品(批号:201230601)约25 mg,精密称定,共5份,分别置于25 ml量瓶中:1份加入2 mol/L盐酸溶液1 ml,放置1 h;1份加入30%过氧化氢溶液1 ml,放置2 h;1份加入流动相1 ml使溶解,在强光4 500 lx下照射4 h;1份加入流动相1 ml使溶解,并在100 ℃水浴中加热2 h;1份加入2 mol/L氢氧化钠溶液1 ml,放置1 h。上述5份溶液分别用流动相B稀释至刻度,摇匀,各量取10 μl,分别注入HPLC仪,记录色谱,详见图3。结果表明,样品在酸、碱、氧化、高温和光照条件下均保持相对稳定,各破坏条件下产生的降解产物峰与主峰均能达到基线分离,且各杂质间的分离度良好。
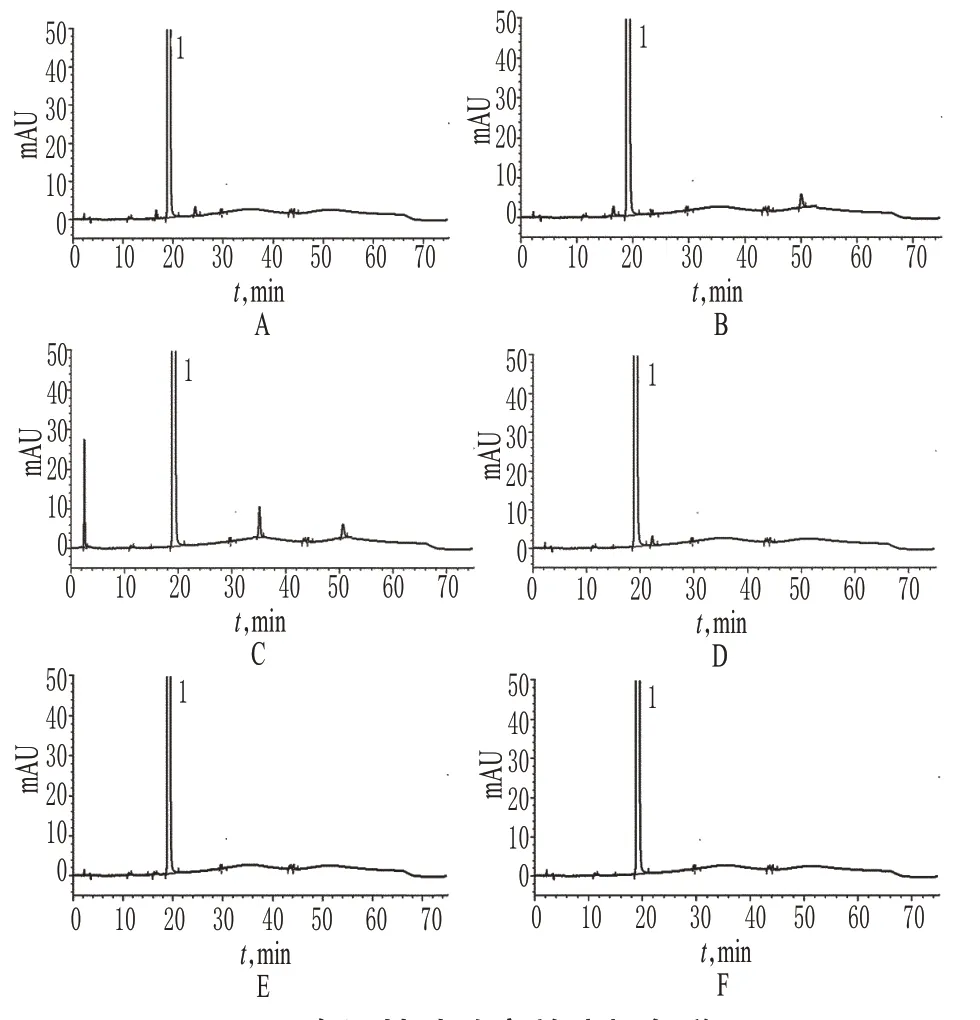
图3 专属性试验高效液相色谱图A.酸破坏样品;B.碱破坏样品;C.氧化破坏样品;D.高温破坏样品;E.光照破坏样品;F.未破坏样品;1.盐酸替罗非班Fig 3 HPLC chromatograms of specificity testA.sample destroyed by acid;B.sample destroyed by alkali;C.sample destroyed by oxidation;D.sample destroyed by high temperature;E.sample destroyed by illumination sample;F.undestroyed sample;1.tirofiban hydrochloride;
2.6 线性关系考察
取盐酸替罗非班对照品适量,精密称定,加流动相B溶解并稀释制成每1 ml中约含1 000 μg的线性贮备液;分别取盐酸替罗非班中间体Ⅰ、Ⅱ、Ⅲ适量,精密称定,分别加流动相B溶解并稀释制成每1 ml中各约含100 μg的线性贮备液。分别精密量取上述各线性贮备液适量,用流动相B按逐级稀释的方法制成系列浓度溶液,测定峰面积,以峰面积(y)对质量浓度(x,μg/ml)进行线性回归,回归方程和线性范围见表2。
2.7 检测限和定量限
分别取“2.6”项下盐酸替罗非班中间体Ⅰ、Ⅱ、Ⅲ线性贮备液适量,均采用逐步稀释法稀释至0.05 μg/ml,按“2.1”项下色谱条件进样测定,记录色谱,以信噪比3计算得3个中间体的最低检测限均为0.5 ng;以信噪比10计算得3个中间体的定量限均为2.0 ng。

表2 盐酸替罗非班及其中间体的回归方程和线性范围Tab 2 The regression equation and linear range of tirofiban hydrochloride and intermediates
2.8 精密度、重复性和稳定性试验
取“2.2”项下的盐酸替罗非班对照品溶液,按“2.1”项下色谱条件重复进样6次,记录色谱,结果盐酸替罗非班峰面积的RSD为0.35%,表明仪器精密度良好。取样品(批号:201230601)适量,平行6份,按“2.2”项下方法处理制备含量测定供试品溶液,按“2.1”项下色谱条件进样测定,得平均含量为99.8%,盐酸替罗非班峰面积的RSD为0.19%,表明本方法重复性良好。取“2.2”项下含量测定供试品溶液(批号:201230601)适量,平行6份,分别在室温下放置0、2、4、8、12、24 h后进样测定,计算得到盐酸替罗非班峰面积的RSD为0.33%,表明供试品溶液在24 h内稳定性良好。
2.9 回收率试验
取盐酸替罗非班对照品约8、10、12 mg,各3份,精密称定,分别置于100 ml量瓶中,加流动相B使其溶解,定容,摇匀,进样测定,记录色谱,计算回收率,结果详见表3。
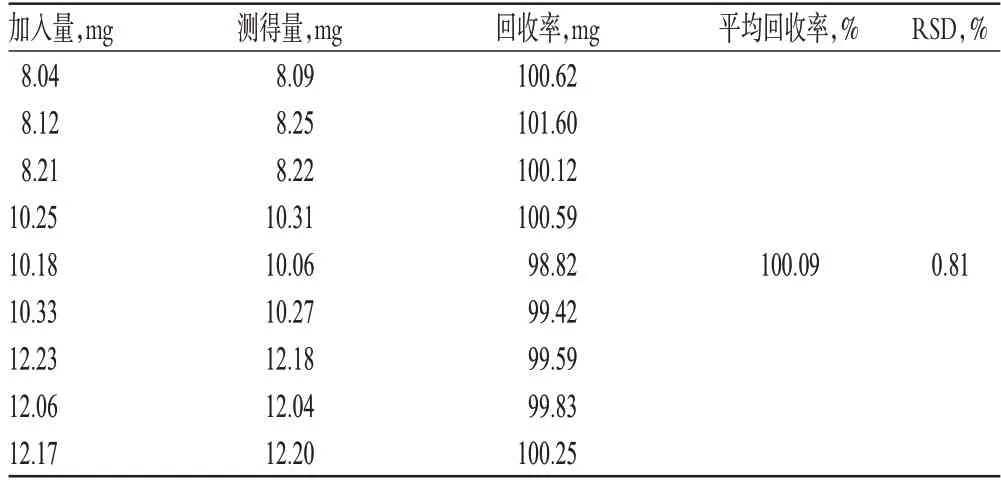
表3 回收率试验结果(n=3)Tab 3 Results of recovery test(n=3)
2.10 样品测定
取对照品和3批样品适量,按“2.2”项下方法处理并按“2.1”项下色谱条件进样测定,计算含量;取3批样品适量,按“2.3”项下方法处理并进样测定,计算有关物质的量(各单个杂质均不得过0.2%,总杂质不得过0.6%),结果见表4。
3 讨论

表4 样品测定结果(n=3)Tab 4 Results of sample determination(n=3)
3.1 流动相的选择[4-5]
本试验在筛选流动相过程中,曾设计了多组不同组成的流动相体系(磷酸盐-乙腈,辛烷/磷酸盐-乙腈,醋酸盐-甲醇,醋酸盐-甲醇/乙腈等),通过对每个组成的流动相体系进行考察,发现醋酸铵-甲醇/乙腈体系按本试验比例混合后能满足分析要求。同时,缓冲液的pH对峰形和分离度也有较大的影响:随着pH的降低,盐酸替罗非班与各中间体及杂质的分离度增加,且峰形较尖锐。但考虑到色谱柱的耐酸性及使用寿命,最终确定流动相pH为2.30。
3.2 梯度洗脱程序的确定[6-7]
盐酸替罗非班与其3个中间体结构较相似,出峰时间较接近,因此选择合适的梯度洗脱方式才能达到分离度要求。中间体Ⅰ与中间体Ⅲ相邻较近,难以分开,故将此处的梯度变缓;而盐酸替罗非班与中间体Ⅱ之间没有其他杂质,故调节此处梯度变化,使中间体Ⅱ快速出峰,以节省洗脱时间。通过对梯度的调整,在表1所示梯度洗脱程序下,主成分与各中间体、杂质均能达到有效分离,且各峰纯度都达100%。故选择此梯度洗脱程序进行洗脱。
3.3 色谱柱的选择[8-10]
在选定流动相系统下,对CAPCELL柱、Thermo和Varian键合相柱进行比较,结果发现,采用Thermo键合相柱分析时,中间体Ⅰ与中间体Ⅲ不能实现良好分离;而采用Varian键合相柱分析时峰形较差,且流动相的pH接近其耐酸值极限。故最终采用CAPCELL PAK C18为本研究的色谱柱。
综上所述,该方法简便、准确、专属性强、灵敏度高,适用于盐酸替罗非班原料药的含量及有关物质测定。
[1]Zhu TQ,Zhang Q,Ding FH,et al.Randomized comparison of intracoronary tirofiban versus urokinase as an adjunct to primary percutaneous coronary intervention in patients with acute ST-elevation myocardial infarction:results of the ICTUS-AMI trial[J].Chin Med J:Engl,2013,126(16):3 079.
[2]Kellert L,Hametner C,Stampfl S.Response to letter regarding article,endovascular stroke therapy:tirofiban is associated with risk of fatal intracerebral hemorrhage and poor outcome[J].Stroke,2013,44(9):e113.
[3]吴依风.盐酸替罗非班治疗急性冠脉综合征80例临床观察[J].中国现代药物应用,2009,3(8):114.
[4]郝玲花,戚燕,杜冠华,等.HPLC法测定布洛芬注射液的有关物质[J].中国新药杂志,2013,22(12):1 396.
[5]赵喆,唐素芳.HPLC法测定地奥司明原料药及片剂的含量和有关物质[J].中国药房,2013,24(29):2 773.
[6]李春艳,张文胜,康仪,等.HPLC测定HX0969w原料药的含量及有关物质[J].华西药学杂志,2013,28(3):300.
[7]张西如,高燕霞,姜建国.洛伐他汀胶囊有关物质的HPLC测定[J].中国药师,2009,12(11):1 659.
[8]洪建文,李趣嫦,王彦蝶.HPLC法测定阿莫西林颗粒剂的含量及有关物质[J].广东药学院学报,2009,25(1):42.
[9]林洁,程煜凤,康秋梅,等.奥氮平有关物质的HPLC检查法研究[J].中国测试,2013,39(4):61.
[10]李洁.地塞米松磷酸钠注射液有关物质检查方法的探讨[J].中国药事,2013,27(4):416.
猜你喜欢
煤化工(2022年3期)2022-07-08
中国药学药品知识仓库(2022年10期)2022-05-29
健康体检与管理(2022年4期)2022-05-13
色谱(2021年7期)2021-06-07
中华养生保健(2020年10期)2021-01-18
汕头大学学报(自然科学版)(2020年4期)2020-12-14
首都食品与医药(2020年1期)2020-10-21
世界最新医学信息文摘(2020年19期)2020-03-31
中国现代药物应用(2017年12期)2017-07-18
中成药(2017年5期)2017-06-13
