
帕唑帕尼衍生物的合成与初步的活性评价
2015-03-13 07:36吴智鸿赵砚瑾王永珍姚郑林江发龙匡先照张养军李庶心
化学工程师 2015年4期
吴智鸿,赵砚瑾,王永珍,姚郑林,江发龙,匡先照,张养军,李庶心
(1.广西医科大学药学院,广西南宁530021;2.军事医学科学院放射与辐射医学研究所,北京100850)
帕唑帕尼(pazopanib,Votrient)是英国葛兰素史克(GSK)公司研发的一种口服VEGF-2 抑制剂,通过抑制VEGFR 自身的磷酸化,使VEGFR 酪氨酸激酶失活,阻断VEGF 信号[1,2]在肿瘤细胞中的表达,从而抑制血管生成,达到“饿死”肿瘤的目的。已于2009年10月获美国FDA 批准上市,用于治疗晚期肾细胞癌[3,4]。临床试验显示其对肾细胞癌、肉瘤等多种肿瘤有抑制作用,口服生物利用度和药代动力学性质较好,不良反应和毒副作用相对较少[5-7]。作为抗肿瘤药物,帕唑帕尼有许多优点:(1)不易耐药,血管内皮细胞基因[8]相对稳定;(2)抗瘤谱广,所有肿瘤都要依赖于血管供给营养[9];(3)副作用少,针对肿瘤血管内皮细胞[10],正常组织血管不易受损;(4)易到达作用部位,药物直接作用于新生血管壁。但其同时仍然存在多种不良反应,最常见的包括腹泻、恶心、呕吐、疲劳、高血压、头发颜色改变和食欲减退。而蛋白尿、AST 升高、锥体外系失调、静脉血栓、肿瘤出血等也很常见[11,12],限制了临床应用。
本研究的目的是结合帕唑帕尼的构效关系,在保持原有活性必须基团的基础上,对非活性必须基团进行化学修饰,以储备新结构化合物并用于筛选低毒、高效且理化性质更稳定的抗肿瘤化合物。帕唑帕尼类化合物的构效关系[13]为:(1)嘧啶环上的N1、C2可与VEGFR-2 催化位点的Cys919 的多肽骨架酰胺形成氢键相互作用,为活性必须。嘧啶环与吲唑环间的氮原子甲基化对活性影响不大,只会改变口服生物利用度和药代动力学性质;(2)吲唑环除了改进该类化合物的药代动力学性质(包括体内清除率和生物利用度)外,芳香化的吲唑环的π 电子云与Lys868 的ε 氨基可产生较强的相互作用;3-甲基吲唑环上的N 对细胞色素P450 酶(CYP450)的血红素铁有很强的结合作用,导致对大量CYP450 同功酶有抑制性等副作用。为了降低该副作用,可在N 上引入一些取代基增加位阻;(3)苯磺酰胺基团同样是其活性必不可少的片段。
依据帕唑帕尼的构效关系,本研究尝试做出了如下修饰:在嘧啶环C5进行氯取代;去掉嘧啶环与吲唑环间的氮原子上的甲基;对换苯磺酰胺与吲唑环的位置;丰富吲唑环的结构。并以3-硝基-6-甲基苯胺和3-硝基-6-乙基苯胺为原料,通过成环、氨基甲基化、硝基还原、亲核取代反应合成得到了3个目标化合物(如图1),并通过了1H NMR 和LC-MS 的确证,且均未见文献报道。合成路线步骤少,产率高易纯化,可为同类型化合物的合成提供参考。
1 实验部分
1.1 仪器与试剂
JNM-ECA-400 超导核磁仪(400MHz日本电子株式会社);API-150ex(ESI)型质谱仪(美国AB 公司)。
3-硝基-6-甲基苯胺、3-硝基-6-乙基苯胺和2,4,5-三氯嘧啶,北京偶合有限公司,其余试剂均为国产化学纯或分析纯。
1.2 目标化合物的合成路线
目标化合物1~5 的合成路线见图2。
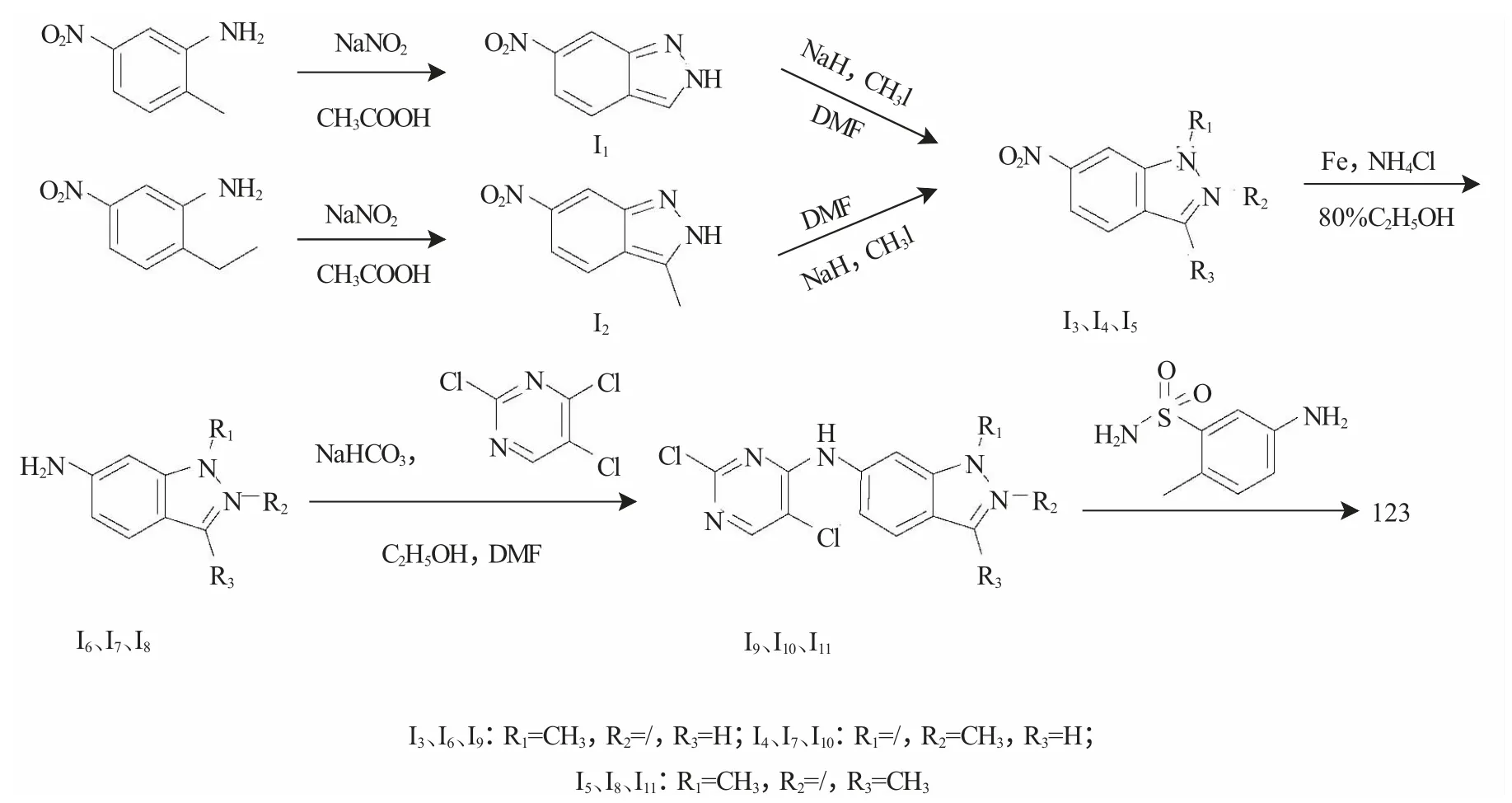
图2 目标化合物的合成路线Fig.2 Synthetic route of the target compounds
1.3 中间体的合成
1.3.1 6-硝基-2H-吲唑(I1) 将1g 3-硝基-6-甲基苯胺(6.6mmol),加入到20mL HAc 中,再分批加入0.57g NaNO2(8.3mmol),常温搅拌0.5h 后,TLC显示反应完全,减压蒸干溶剂,用乙酸乙酯(30mL)溶解,再用饱和NaHCO3(20mL×3)洗涤,有机层用无水MgSO4干燥,抽滤,减压浓缩,烘干后得橘黄色固体0.5g,产率为47.3%。
1.3.2 3-甲基-6-硝基-2H-吲唑(I2) 实验方法同1.3.1 项下,主要原料投料量分别为1g 3-硝基-6-乙基苯胺(6.0mmol),NaNO2(0.52g,7.57mmol),得棕黄色固体0.47g,产率为44.3%。
1.3.3 1-甲基-6-硝基-1H-吲唑(I3) 在100mL三颈瓶中,加入20mL 重蒸DMF,再分批加入0.59g NaH(24.6mmol),冰浴条件下搅拌10min,将2g 6-硝基-2H-吲唑(12.3mmol)分批加入反应瓶中,15min 后恢复室温反应,常温搅拌0.5h 后,滴加CH3I 2.9g(20.4mmol),滴毕搅拌10min,TLC 显示反应完全,将反应液倒入200mL 水中,有固体析出,过滤,滤液用乙酸乙酯萃取(20mL×2),有机层用无水MgSO4干燥,抽滤,减压浓缩,与滤饼一起烘干,通过干法过层析柱得固体0.95g。产率为43.8%。
1.3.4 2-甲基-6-硝基-2H-吲唑(I4) 实验方法同1.3.3 项下,主要原料投料量分别为NaH(0.59g,24.6mmol),6-硝基-2H-吲唑(2g,12.3mmol),CH3I 2.9g(20.4mmol),得1.15g 固体,产率为52.8%。
1.3.5 1,3-二甲基-6-硝基-1H-吲唑(I5) 实验方法同1.3.3 项下,主要原料投料量分别为NaH(0.27g,11.3mmol),3-甲基-6-硝基-2H-吲唑(1g,5.6 mmol),CH3I(2.4g,16.9mmol),得固体0.68g,产率为63.5%。
1.3.6 1-甲基-6-胺基-1H-吲唑(I6) 在100mL三颈瓶中,加入1-甲基-6-硝基-1H-吲唑1g(5.6 mmol),Fe 粉2.5g(44.6mmol),NH4Cl 1.2g(22.4mmol),乙醇/ 水(4∶1)20mL,搅拌条件下慢慢升温至80℃,0.5h 后,TLC 显示反应完全,趁热过滤,滤液减压浓缩,乙酸乙酯(30mL)溶解,再用清水(20mL×3)洗涤,有机层用无水硫酸镁干燥,抽滤,减压浓缩,烘干后得土黄色固体0.75g,产率为90.3%。
1.3.7 2-甲基-6-胺基-2H-吲唑(I7) 实验方法同1.3.6 项下,主要原料投料量分别为2-甲基-6-硝基-2H-吲唑(3g,16.9mmol),Fe 粉(7.6g,135.7 mmol),NH4Cl(2.7g,50.4mmol),得土黄色固体1.5g,产率为60%。
1.3.8 1,3-二甲基-6-胺基-1H-吲唑(I8) 实验方法同1.3.6 项下,主要原料投料量分别为1,3-二甲基-6-硝基-1H-吲唑(4g,20.9mmol),Fe 粉(9.4g,167.9mmol),NH4Cl(3.4g,63.5mmol),得固体2.6g,产率为76.7%。
1.3.9 (2,5-二氯-嘧啶-4-基)-(1-甲基-1H-吲哚-6-基)-胺(I9) 将1g1-甲基-6-胺基-1H-吲唑(6.8mmol),0.3g NaHCO3(3.6mmol)溶于20mL 乙醇及5mLTHF 中,加入2,4,5-三氯嘧啶3.7g(20.2mmol),升温至75℃,反应4h 后,有固体析出,TLC 显示反应完全,过滤滤饼用乙醇(10mL×2)洗涤,烘干得黄白色固体0.67g,产率为33.5%。
1.3.10 (2,5-二氯-嘧啶-4-基)-(2-甲基-2H-吲哚-6-基)-胺(I10) 实验方法同1.3.9 项下,主要原料投料量分别为2-甲基-6-胺基-2H-吲唑(1.5g,10.2mmol),NaHCO3(0.43g,5.1mmol),2,4,5-三氯嘧啶(5.6g,30.6mmol),得灰白色固体1g,产率为34.0%。
1.3.11 (2,5-二氯-嘧啶-4-基)-(1,3-二甲基-1H-吲哚-6-基)-胺(I11) 实验方法同1.3.9 项下,主要原料投料量分别为1,3-二甲基-6-胺基-1H-吲唑(1g,6.2mmol),NaHCO3(0.26g,3.1mmol),2,4,5-三氯嘧啶(3.4g,18.6mmol),得灰白色固体0.62g,产率为32.5%。
1.4 目标化合物的合成
1.4.1 目标化合物1 将0.4g(2,5-二氯-嘧啶-4-基)-(1-甲基-1H-吲哚-6-基)-胺(0.9mmol)与0.25g5-氨基-2-甲基-苯磺酰胺(1.3mmol)溶于20mL 异丙醇中,加入1mL 浓HCl,升温至回流14h 后,TLC 显示反应完全,过滤,滤饼用乙醇(5mL×3)洗涤,烘干得白色固体0.32g,产率为60.0%。
1.4.2 目标化合物2 实验方法同1.4.1 项下,主要原料投料量分别为(2,5-二氯-嘧啶-4-基)-(2-甲基-2H-吲哚-6-基)-胺(0.4g,1.36mmol),5-氨基-2-甲基-苯磺酰胺(0.25g,1.3mmol),得白色固体0.29g,产率为50.3%。
1.4.3 目标化合物3 将(2,5-二氯-嘧啶-4-基)-(1,3-二甲基-1H-吲哚-6-基)-胺(0.4g,1.3mmol)与5-氨基-2-甲基-苯磺酰胺(0.25g,1.3mmol)溶于20mL 仲丁醇中,加入1mL CF3COOH,升温至回流40h 后,TLC 显示反应完全,过滤,滤饼用乙醇(5mL×3)洗涤,烘干得白色固体0.25g,产率为42.0%。
1.5 化合物的体外活性初步评价实验
1.5.1 实验原理 MTT 分析法是以活细胞代谢物还原剂噻唑蓝(MTT)为基础的分析方法。噻唑蓝可作用于活细胞的线粒体中的呼吸链,在琥珀酸脱氢酶和细胞色素C 的作用下四唑环开裂,生成蓝色的甲瓒结晶,甲瓒结晶的生成量仅与活细胞的数目成正比(死细胞中琥珀酸脱氢酶消失,不能将噻唑蓝还原)。甲瓒结晶可被二甲基亚砜(DMSO)溶解,利用酶标仪测定570nm 处的光密度(OD)值,可以反映出活细胞的数目。
1.5.2 实验方法 取培养4~5d,并处于对数生长期的胃细胞癌MGC803 细胞,制成细胞悬液计数。用完全培养基RPMI1640 稀释细胞悬液至所需的细胞浓度。取96 孔平板,每孔加细胞悬液180μL,接种细胞3000/孔,加细胞过程控制在4h,于37℃、5%CO2温箱孵育24h。受试化合物分别溶解并依次稀释成5 个浓度(10-5,5×10-6,10-6,10-7和10-8mol·L-1),每孔加入20μL,在37℃、5%CO2及100%湿度,孵育4~5d。噻唑蓝用生理盐水配成5mg·mL-1溶液,每孔加20μL,37℃温育4h,使噻唑蓝还原为甲臜。吸出上清液,加入200μL 的二甲基亚砜(DMSO)使甲臜溶解,用平板摇床摇匀。用自动化分光光度平板读数计(Model 312,Biotech Research Laboratories,Inc.,Rockville,Md)在570nm 处检测每个小孔的光密度。
2 结果
2.1 化合物的结构鉴定
目标化合物1: 白色固体,ESI-MS m/z:444.1[M+H]+,分子式为C19H18ClN7O2S。1H NMR(400MHz,DMSO)δ:2.51(s,3H),3.94(s,3H),6.93-6.95(d,1H),7.32-7.39(m,3H),7.74-7.76(d,1H),7.79-7.82(dd,1H),7.93(s,2H),8.032-8.034(d,1H),8.30(s,1H),9.51(s,1H),10.10(s,1H).
目标化合物2: 白色固体,ESI-MS m/z:444.1[M+H]+,分子式为C19H18ClN7O2S。1H NMR(400MHz,DMSO)δ:2.51(s,3H),4.17(s,3H),7.02-7.04(d,1H),7.24-7.26(dd,1H),7.39(s,2H),7.69-7.71(d,1H),7.78-7.81(dd,1H),7.89-7.92(d,2H),8.29(s,1H),8.35(s,1H),9.52(s,1H),10.16(s,1H).
目标化合物3: 白色固体,ESI-MS m/z:458.1[M+H]+,分子式为C20H20ClN7O2S。1H NMR(400MHz,DMSO)δ:3.85(s,3H),4.72-4.77(t,6H),6.98-7.00(d,1H),7.29-7.33 (m,3H),7.66-7.68(d,1H),7.82-7.88(m,3H),8.23(s,1H),9.27(s,1H),9.82(s,1H).
2.2 化合物的初步活性评价
初步计算的结果显示,目标化合物1~3 对MGC803 细胞的IC50分别为(0.868±0.096)、(88.805±10.819)、(4.322±1.790)μmol·L-1,阳性对照帕唑帕尼的IC50为(4.436±0.757)μmol·L-1。表明化合物1、3 的活性超过或接近阳性对照物帕唑帕尼,表明它们具有较高的抗肿瘤活性,其中1 的生物活性为帕唑帕尼的5 倍,可选为先导化合物作进一步研究,为找寻新的具有更高活性的先导化合物提供了可能。
3 讨论
有文献[5]报道在制备2,3-二甲基-6-硝基-2H-吲唑的过程中,需要用到的原料是三甲基氧鎓四氟硼酸盐或是硫酸二甲酯,但是前者价格昂贵,后者污染严重。本文参考此法,改进反应条件,成功地以氢化钠和碘甲烷为原料,合成出了1,3-二甲基-6-硝基-1H-吲唑(I5)。虽然产率由70%略微降到了63.5%,但实验更经济环保。
目标化合物的体外活性测试表明:(1)1H-吲唑环相比2H-吲唑环有更高的活性,两个活性较好的化合物均为1H-吲唑结构。(2)在嘧啶环C-5 位引入卤素如:氯,有利于增强本类化合物对靶酶的抑制活性。(3)对比目标化合物1、3,表明在吲唑环上即使有相同的取代基,但其余的取代基不同,也会导致化合物的活性有一定的差别。
本文以葛兰素史克公司研发的多靶点VEGFR络氨酸激酶抑制剂帕唑帕尼为先导化合物,寻找合理的构建单元,设计合成了3 个帕唑帕尼衍生物。其中1、3 具有较高的抗肿瘤活性,为找寻低毒、高效、稳定的抗肿瘤药物提供了可能。
[1]Friesen D Y,Lorenz D A,Smith S W. Pharmaceutical compostions comprising an amorphous form ofa VEGF-R inhibitor[P]. WO:2006123223,2006-11-23.
[2]Babu S,Dagnino R J,Ouellette M,et al.Methods for preparing indazole compounds[P].WO:2006048745,2006-05-11.
[3]Harris PA,BOLOOR A,CHEUNG M,et al. Discovery of 5-[[4-[2,3-dimethyl-2H-indazol-6-yl)methylamino]-2-pyrimidinyl]amino]-2-methyl-benzenesulfonamide (pazopanib),a nobel and potent vascular endothelial growth factor receptor inhibitor[J]. J Med.Chem.,2008,51(7):4632-4640.
[4]陈燕,方正,韦萍.盐酸帕唑帕尼的合成[J].中国医药工业杂志,2010,41(5):326-328.
[5]Sloan B,Scheinfeld NS. Pazopanib,a VEGF receptor tyrosine kinase inhibitor for cancer therapy[J].Curr Opin InvestigDrugs,2008,9(12):1324-1335.
[6]Sonpavde G,Hutson TE,Sternberg CN. Pazopanib,a potent orally administered small-molecule multitargeted tyrosine kinase inhibitor for renal cell carcinoma[J]. Expert Opin Investig Drugs,2008,17(2):253-261.
[7]郑宇静,姜文亮,王志宏,等.酪氨酸激酶抑制剂帕唑帕尼的药理与临床研究[J].中国新药杂志,2011,20(12):1057-1060.
[8]祁浩飞,王兵,刘冰妮,等.帕唑帕尼盐酸盐的合成[J].中国现代应用药学,2011,28(1):58-60.
[9]祖强,洪宝发,符伟军,等.分子靶向药物Pazopanib 治疗转移性肾癌的效果观察[J].现代泌尿外科杂志,2008,13(4):258-260.
[10]Deininger M,Buchdunger E,Druker BJ.The development of imatinib as a therapeutic agent for chronic myeloid[J].B lood,2005,105(7):2640-2653.
[11]HURWITZ HI,DOWLATI A,SAINI S,et al.Phase I trial of pazopanib(GW786034),an oral multikinase angiogenesis inhibitor,in patients with advanced cancer:results of safety,pharmacokinetics,and clinical activity[J]. Clin Cancer Res,2009,15(12):4220-4227.
[12]LAPLANT KD. Pazopanib: an oral multitargeted tyrosine kinase inhibitor for use in renal cell carcinoma[J]. Ann Pharmacother,2010,44(6):1054-1060.
[13]杨欣,唐家邓,岑均达.盐酸帕唑帕尼的合成[J].中国医药工业杂志,2012,(08):644-646.
猜你喜欢
昆明医科大学学报(2021年8期)2021-08-13
化工管理(2021年7期)2021-05-13
上海计量测试(2020年1期)2020-03-18
武警医学(2018年10期)2018-11-06
中国农资(2016年1期)2016-12-01
浙江大学学报(工学版)(2016年2期)2016-06-05
当代化工研究(2016年6期)2016-03-20
环境科技(2015年1期)2015-11-08
火炸药学报(2014年5期)2014-03-20
无机化学学报(2014年8期)2014-02-28
