
硝基甲烷在石墨烯表面初始反应机理的理论研究
2015-05-10 02:21刘英哲来蔚鹏葛忠学
含能材料 2015年9期
刘英哲, 康 莹, 来蔚鹏, 尉 涛, 葛忠学
(西安近代化学研究所, 陕西 西安 710065)
1 引 言
基于碳纳米材料掺杂的复合含能材料能够改善传统含能材料的某些性能,如在HMX中添加C60、石墨烯(Graphene, GRA)氧化物等碳纳米材料能够有效降低HMX的感度,并增加了热解反应的活化能,从而提高了热分解稳定性[1-2]。理论计算[3-4]预测通过碳纳米管封装新型高能量密度物质N8等,能够提高其室温常压下的稳定性。除了新型高能量密度材料,典型含能化合物RDX(hexahydro-1,3,5-trinitro-striazine)、HMX(octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine)、FOX-7(1,1-diamino-2,2-dinitroethylene)等封装在碳纳米管空腔、双层GRA间隙时,其稳定性也能得到一定程度的提高[5]。此外,GRA及其氧化物还能催化硝基甲烷(Nitromethane, NM)的燃烧[6-7]。从微观角度来说,碳纳米材料可能改变了含能材料的结构、能量及反应活化能等,使含能材料的感度、稳定性及反应速率等性质发生了变化,因此研究含能材料在碳纳米材料掺杂时的反应机理及相应的结构与能量变化具有重要的意义。
硝基甲烷(NM)是最简单的含能化合物,常作为模型广泛用于理论研究中,包括结构性质[8-13]、融化与结晶[14-15]、热分解[16-19]等方面。然而,NM的反应机理仍十分复杂,在反应过程中会出现许多基元反应以及新的物质。因此,对于含能材料反应机理的研究通常先集中在初始反应上。NM初始反应包括硝基甲烷-亚硝酸甲酯(methyl nitrite, MN)重排反应、氢迁移重排反应及C—N键均裂反应,反应式分别如下[19]:
CH3NO2→ CH3ONO
(1)
CH3NO2→ CH2NOOH
(2)
CH3NO2→·CH3+·NO2
(3)
尽管碳纳米材料可作为钝感剂、催化剂、添加剂应用在含能材料领域,并且表现出了良好的效果[20],但是碳纳米材料对含能化合物反应机理的影响仍研究不够完全。因此,本研究选取GRA与NM作为碳纳米材料与含能化合物的模型,采用组合量子化学ONIOM方法探索GRA对NM初始反应、结构与能量的影响。
2 计算方法
2.1 分子模型
单层GRA初始结构由VMD软件[21]中Carbon Nanostructure Builder模块构建,尺寸为20Å× 20Å,共含有180个碳原子,其边缘的悬键用38个氢原子进行饱和。NM有交错式和重叠式两种异构体,H—C—N—O二面角最小值分别为30°和0°,初始结构由VMD软件[21]中Molefacture模块构建。
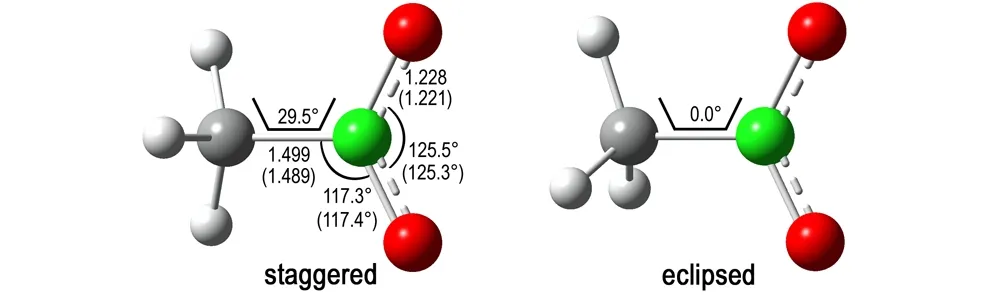
图1NM在B3LYP/6-31+G**理论水平下的优化结构(括号内数据为实验值[22],键长单位为Å)
Fig.1The optimized geometries of NM at B3LYP/6-31+G**level (data in parentheses are the experimental values[22], the bond length unit is given in Å)
2.2 量化计算
考虑到研究体系共含有225个原子,难以完全用高精度的量子化学方法进行计算,故采用组合量子化学ONIOM(our Own N-layer Integrated molecular Orbital and molecular Mechanics)方法[23]将体系分为不同计算精度的两层进行处理: 内层为NM,采用高精度的密度泛函理论B3LYP/6-31+G**[24-25]进行计算; 外层为GRA,采用分子力学方法UFF力场[26]进行计算。原子电荷由Qeq方法[27]计算,利用电子内嵌技术[23]描述内层与外层之间的静电相互作用。为了确认最稳定的几何构型与反应过渡态,在ONIOM (B3LYP/6-31+G**:UFF)理论水平下对振动频率进行求解。利用内禀反应坐标法[28]进一步确认反应路径。作为对比,采用B3LYP/6-31+G**对孤立NM进行同样的计算。所有理论计算均采用Gaussian09[29]软件完成。
3 结果与讨论
3.1 结构分析
采用B3LYP/6-31+G**理论水平计算的NM最优结构与实验结果[22]吻合较好,仅C—N键比实验值长0.01Å(见图1)。重叠式与交错式NM之间的能量差小于0.05 kJ·mol-1,表明—CH3与—NO2基团几乎能够绕着C—N键自由地旋转,这与文献[30]理论计算结果是一致的。然而,NM在GRA表面的最优结构基本保持交错式的构型,不论NM初始结构为重叠式还是交错式,此时NM能够最大程度地与GRA平面结构相互作用(见图2)。这与NM在另一种碳纳米材料——碳纳米管中的结构有所不同,量子化学计算表明NM在(5,5)碳纳米管空腔内的最优结构接近于重叠式构型[31],表明碳纳米材料的几何形状对NM的最优结构有着直接影响。与孤立交错式结构相比,NM在GRA表面的主要结构变化为H—C—N—O二面角从29.5°(见图1)增加到30.9°(见图2)。另外,NM距GRA表面约3Å,处于合理的非键作用区域。
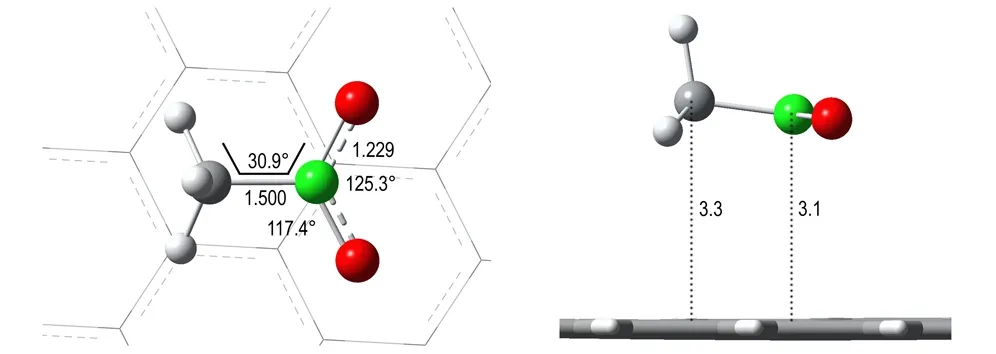
图2NM@GRA在ONIOM (B3LYP/6-31+G**:UFF)理论水平下的优化结构
Fig.2The optimized geometries of NM@GRA at ONIOM (B3LYP/6-31+G**:UFF) level
3.2 NM-MN重排反应
对于NM和NM@GRA,分别在B3LYP/6-31+G**和ONIOM (B3LYP/6-31+G**: UFF)理论水平下计算了NM-MN重排反应中的过渡态,记为TS1和TS1@GRA。图3为NM与NM@GRA在NM-MN重排反应中过渡态及反应产物的优化结构。表1为NM与NM@GRA体系相关结构的总能量、相对能量及振动频率。由图3可以看出,TS1结构中C—N键长为1.977Å, C—O键长为2.029Å,与文献[32]的理论计算结果C—N键长1.9Å和C—O键长2.0Å相似。相比之下,GRA对TS1结构的影响主要体现为: C…O距离由原来的2.029Å增加到2.055Å,同时虚频减小了27.1 cm-1(见表1)。另外,MN在GRA表面构型发生了翻转,—CH3基团围绕C—O键旋转了约30°,即从重叠式转为交错式。究其原因,是由于GRA平面结构的特点所致,MN为交错式时,能够最大程度地与GRA作用。
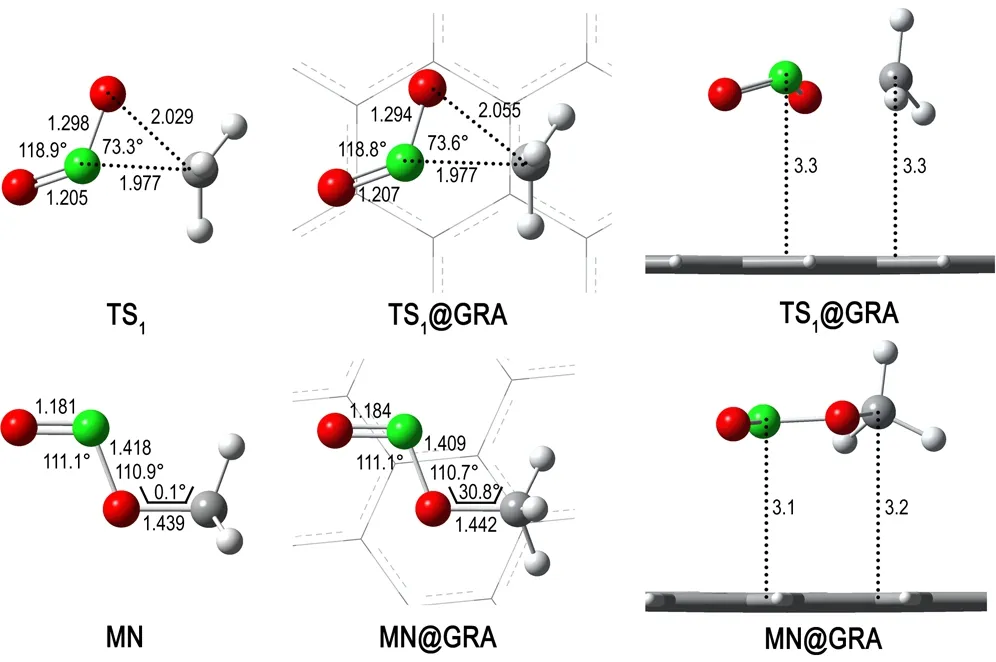
图3NM与NM@GRA在NM-MN重排反应中过渡态及反应产物的优化结构
Fig.3Optimized structures of the transition states and products for the NM-MN rearrangement reaction of NM and NM@GRA
表1NM与NM@GRA体系的总能量、相对能量及振动频率
Table1Total energies, relative energies and vibrational frequencies of NM and NM@GRA systems
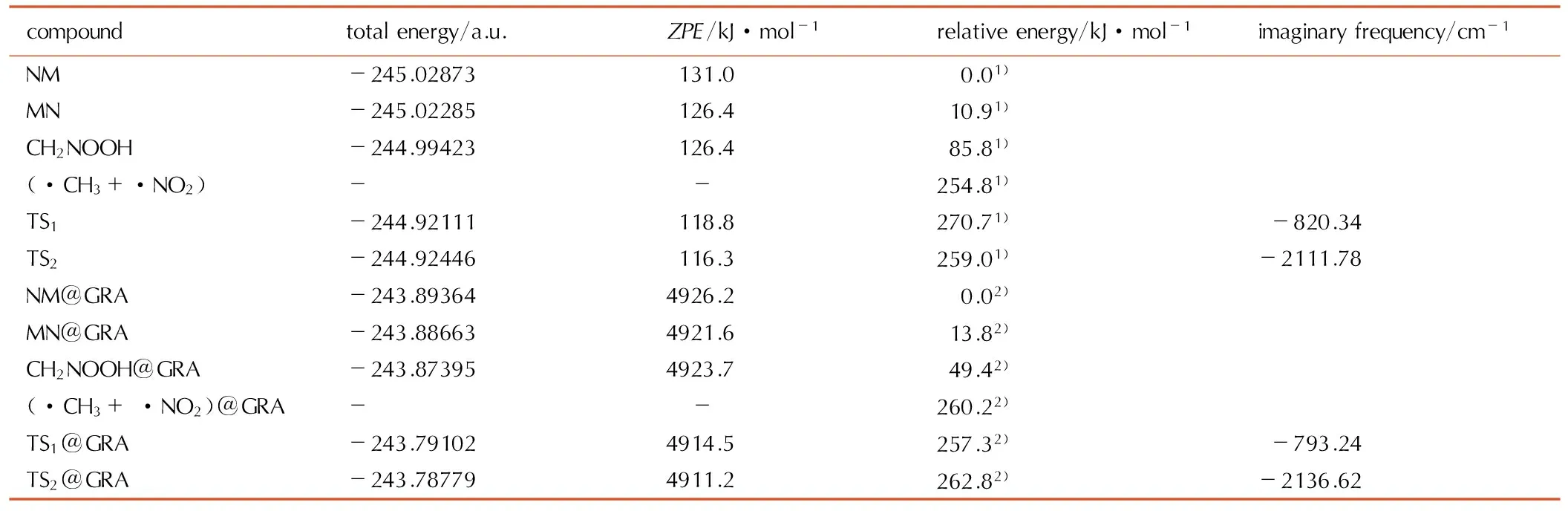
compoundtotalenergy/a.u.ZPE/kJ·mol-1relativeenergy/kJ·mol-1imaginaryfrequency/cm-1NM-245.02873131.0 0.01)MN-245.02285126.410.91)CH2NOOH-244.99423126.485.81)(·CH3+·NO2)- -254.81)TS1-244.92111118.8270.71)-820.34TS2-244.92446116.3259.01)-2111.78NM@GRA-243.893644926.2 0.02)MN@GRA-243.886634921.613.82)CH2NOOH@GRA-243.873954923.749.42)(·CH3+·NO2)@GRA- -260.22)TS1@GRA-243.791024914.5257.32)-793.24TS2@GRA-243.787794911.2262.82)-2136.62
Note: 1) Relative to the NM calculated at the B3LYP/6-31+G**level of theory, 2) Relative to the NM@GRA calculated at the ONIOM(B3LYP/6-31+G**:UFF) level of theory.
由表1可以看出,TS1相对于NM的能量为270.7 kJ·mol-1,这与B3LYP/6-311++G**理论水平计算的271.5 kJ·mol-1 [31]及G2MP2理论水平计算的270.3 kJ·mol-1 [33]十分接近,说明B3LYP/6-31+G**方法是较为可靠的。TS1@GRA相对于NM@GRA的能量为257.3 kJ·mol-1,比TS1降低了约13.4 kJ·mol-1,说明NM-MN重排反应在GRA表面更容易发生。另一方面,MN比NM能量高10.9 kJ·mol-1,MN@GRA比NM@GRA能量高13.8 kJ·mol-1,NM-MN重排的正、逆反应活化能相差不大,表明NM-MN重排反应也能够可逆地发生。
3.3 氢迁移重排反应
氢迁移重排反应是NM-MN重排反应的一个竞争反应,由NM分子内C原子上的一个H原子迁移到O原子上,从而生成CH2NOOH。采用与NM-MN重排反应相同的理论水平,计算NM及NM@GRA氢迁移重排反应的过渡态,记为TS2和TS2@GRA。如图4所示,TS2为四元环过渡态,其中,C…H距离为1.478Å,O…H距离为1.245Å。当NM在GRA表面发生氢迁移重排反应时,TS2@GRA 的结构与TS2相比无明显变化。相反,GRA对产物CH2NOOH的结构有着较大影响,GRA的平面结构诱导CH2NOOH分子也呈平面结构,从而更有利于非键相互作用。在孤立CH2NOOH分子内,与O原子相邻的H原子并不处于其他原子所形成的平面内,H—O—N—C二面角为38.4°,而在CH2NOOH@GRA结构中,该二面角为178.4°。此外,在GRA作用下,CH2NOOH分子内N—O键长增加了0.022Å,N—OH键长缩短了0.023Å。
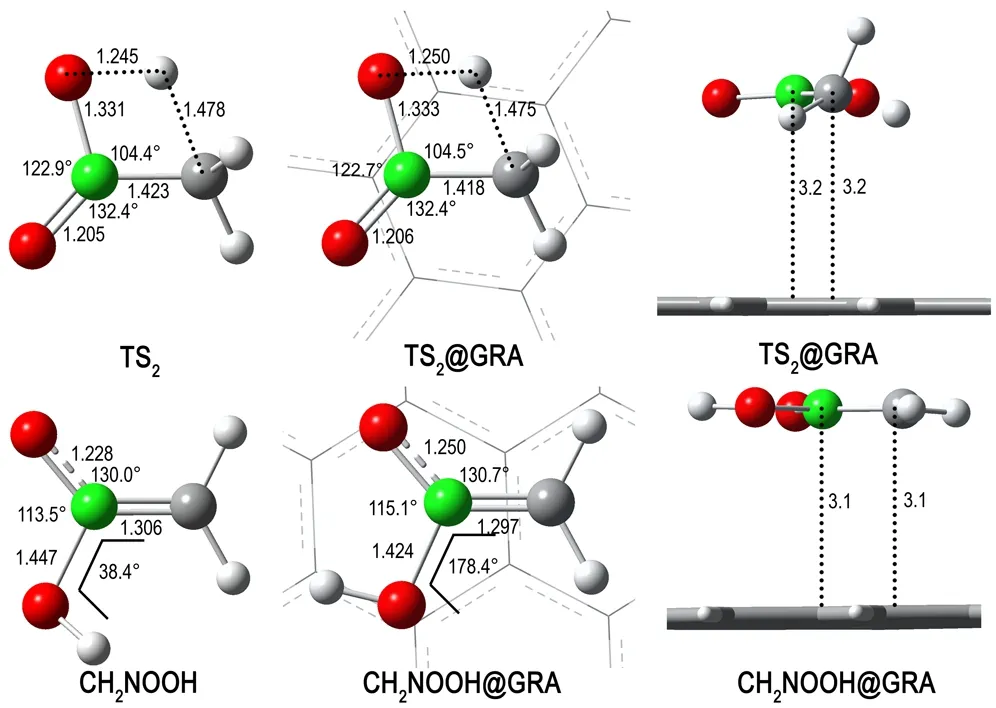
图4NM与NM@GRA在氢迁移重排反应中过渡态及反应产物的优化结构
Fig.4Optimized structures of the transition states and products for the H-migration rearrangement reaction of NM and NM@GRA
TS2相对于NM的能量为259.0 kJ·mol-1(见表1),分别比B3LYP/6-311++G**及G2MP2理论水平计算的结果少0.8 kJ·mol-1 [31]和8.8 kJ·mol-1 [33]。TS2@GRA相对于NM@GRA的能量为262.8 kJ·mol-1,与TS2相差不到4 kJ·mol-1,说明NM在GRA表面发生氢迁移重排反应时反应能垒变化不大。但是,反应产物CH2NOOH在GRA表面上更加稳定,其相对能量比孤立状态时下降了36.4 kJ·mol-1,这源于CH2NOOH分子在GRA诱导下呈平面结构所致。
3.4 C—N键均裂反应
C—N键均裂反应被认为是NM热解反应的主要过程,该反应生成一个甲基与一个硝基的自由基。分别在UB3LYP/6-31+G**和ONIOM (UB3LYP/6-31+G**:UFF)理论水平下扫描了NM与NM@GRA随C—N键变化的势能曲线。为了更好地研究GRA对C—N键均裂反应的影响,在扫描过程中,同时对NM结构进行优化。如图5所示,随着C—N键长的增加,NM与NM@GRA两个体系的能量均增加,并且两个体系在能量上只有微小差距。另外,在C—N键断裂过程中,没有发现过渡态的存在,这与NM在(5,5)碳纳米管内发生C—N键均裂反应时不同[31]: NM在(5,5)碳纳米管内时,由于管内空腔限制,C—N键均裂存在反应过渡态,且反应能垒明显降低。由势能曲线可以得出NM与NM@GRA发生C—N键均裂反应的能垒分别为254.8 kJ·mol-1与260.2 kJ·mol-1,说明NM在GRA表面发生该反应时反应能垒略有增加。
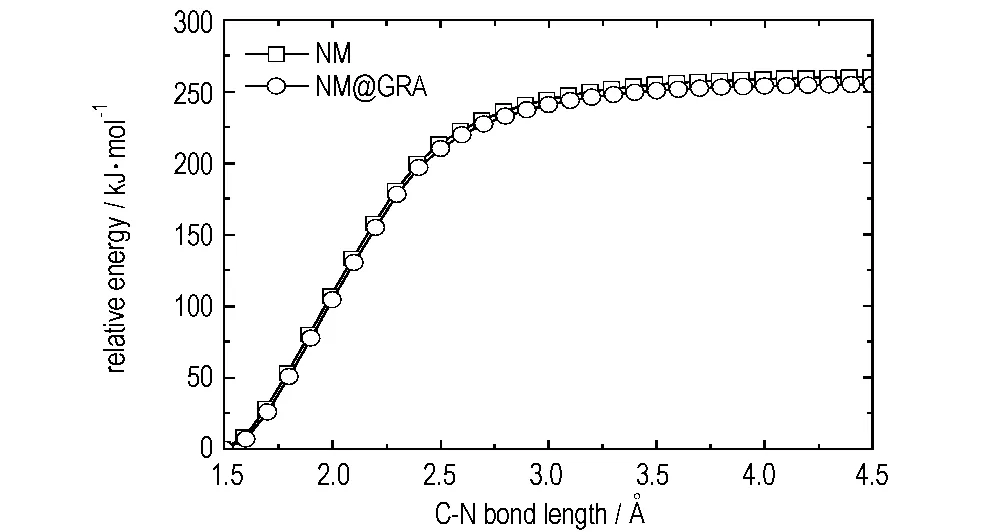
图5NM及NM@GRA发生C—N键均裂反应的势能曲线
Fig.5Potential energy curves of the C—N homolytic cleavage reaction forNM and NM@GRA
3.5 反应势能面
图6为NM与NM@GRA两个体系初始反应的势能面。对于NM,三个初始反应的活化能顺序为C—N键均裂反应<氢迁移重排反应 图6NM(实线)与NM@GRA(虚线)初始反应的势能面曲线(单位为kJ·mol-1) Fig.6Potential energy surface of the initial reaction pathways for NM(solid line) and NM@GRA(dotted line)(relative energies are given in kJ·mol-1) 采用组合量子化学ONIOM方法研究了NM在GRA表面的初始反应,包括NM-MN重排反应、氢迁移重排反应及C—N键均裂反应。计算结果表明,GRA对反应过渡态及产物的结构、能量均有不同程度的影响。GRA表面使NM三种初始反应的活化能依次降低了13.4 kJ·mol-1、增加了3.8 kJ·mol-1及增加了5.4 kJ·mol-1,活化能的顺序由C—N键均裂反应<氢迁移重排反应 参考文献: [1] Jin B, Peng R, Chu S J, et al. Study of the desensitizing effect of different [60]fullerene crystals on cyclotetramethylenetetranitramine (HMX)[J].Propellants,Explosives,Pyrotechnics, 2008, 33(6): 454-458. [2] Li R, Wang J, Shen J P, et al. Preparation and characterization of insensitive HMX/graphene oxide composites[J].Propellants,Explosives,Pyrotechnics, 2013, 38(6): 798-804. [3] Abou-Rachid H, Hu A, Timoshevskii V, et al. Nanoscale high energetic materials: a polymeric nitrogen chain N8confined inside a carbon nanotube[J].PhysicalReviewLetters, 2008, 100(19): 196401. [4] Ji W, Timoshevskii V, Guo H, et al. Thermal stability and formation barrier of a high-energetic material N8polymer nitrogen encapsulated in (5,5) carbon nanotube[J].AppliedPhysicsLetters, 2009, 95(2): 021904. [5] Smeu M, Zahid F, Ji W, et al. Energetic molecules encapsulated inside carbon nanotubes and between graphene layers: DFT calculations[J].JournalofPhysicalChemistryC, 2011, 115(22): 10985-10989. [6] Liu L M, Car R, Selloni, A, et al. Enhanced thermal decomposition of nitromethane on functionalized graphene sheets: ab initio molecular dynamics simulations[J].JournaloftheAmericanChemicalSociety, 2012, 134(46): 19011-19016. [7] Zhang C Y, Wen Y S, Xue X G. Self-enhanced catalytic activities of functionalized graphene sheets in the combustion of nitromethane: molecular dynamic simulations by molecular reactive force field[J].ACSAppliedMaterials&Interfaces, 2014, 6(15): 12235-12244. [8] Alper H E, Abu-Awwad F, Politzer P. Molecular dynamics simulations of liquid nitromethane[J].JournalofPhysicalChemistryB, 1999, 103(44): 9738-9742. [9] Sorescu D C, Rice B M, Thompson D L. Theoretical studies of solid nitromethane[J].JournalofPhysicalChemistryB, 2000, 104(35): 8406-8419. [10] Sorescu D C, Rice B M, Thompson D L. Molecular dynamics simulations of liquid nitromethane [J].JournalofPhysicalChemistryA, 2001, 105(41): 9336-9346. [11] Alavi S, Thompson D L. A molecular-dynamics study of structural and physical properties of nitromethane nanoparticles[J].JournalofChemicalPhysics, 2004, 120(21): 10231-10239. [12] Kabadi V N, Rice B M. Molecular dynamics simulations of normal mode vibrational energy transfer in liquid nitromethane[J].JournalofPhysicalChemistryA, 2004, 108(4): 532-540. [13] Appalakondaiah S, Vaitheeswaran G, Lebègue S. A DFT study on structural, vibrational properties, and quasiparticle band structure of solid nitromethane[J].JournalofChemicalPhysics, 2013, 138(18): 184705. [14] Siavosh-Haghighi A, Thompson D L. Molecular dynamics simulations of surface-initiated melting of nitromethane[J].JournalofChemicalPhysics, 2006, 125(18): 184711. [15] Siavosh-Haghighi A, Sewell T D, Thompson D L. Molecular dynamics study of the crystallization of nitromethane from the melt[J].JournalofChemicalPhysics, 2010, 133(19): 194501. [16] Chang J, Lian P, Wei D Q, et al. Thermal decomposition of the solid phase of nitromethane: ab initio molecular dynamics simulations[J].PhysicalReviewLetters, 2010, 105(18): 188302. [17] Han S P, van Duin A C T, Goddard W A, et al. Thermal decomposition of condensed-phase nitromethane from molecular dynamics from ReaxFF dynamics[J].JournalofPhysicalChemistryB, 2011, 115(20): 6534-6540. [18] Rom N, Zybin S V, van Duin A C T, et al. Density-dependent liquid nitromethane decomposition: molecular dynamics simulations based on ReaxFF[J].JournalofPhysicalChemistryA, 2011, 115(36): 10181-10202. [19] Guo F, Cheng X L, Zhang H. Reactive molecular dynamics simulation of solid nitromethane impact on (010) surfaces induced and nonimpact thermal decomposition[J].JournalofPhysicalChemistryA, 2012, 116(14): 3514-3520. [20] 兰元飞, 李霄羽, 罗运军. 石墨烯在含能材料中的应用研究进展[J]. 火炸药学报, 2015, 38(1): 1-7. LAN Yuan-fei, LI Xiao-yu, LUO Yun-jun. Research progress on application of graphene in energetic materials[J].ChineseJournalofExplosive&Propellants, 2015, 38(1): 1-7. [21] Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics[J].JournalofMolecularGraphics, 1996, 14(1): 33-38. [22] NIST Standard Reference Database Number 69, http://webbook.nist.gov/chemistry/ [23] Vreven T, Byun K S, Komáromi I, et al. Combining quantum mechanics methods with molecular mechanics methods in ONIOM[J].JournalofChemicalTheoryandComputation, 2006, 2(3): 815-826. [24] Becke A D. Density-functional thermochemistry. III. The role of exact exchange[J].JournalofChemicalPhysics, 1993, 98(7): 5648-5652. [25] Lee C, Yang W, Parr R G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density[J].PhysicalReviewB, 1988, 37(2): 785-789. [26] Rappé A K, Casewit, C K, Colwell, K S, et al. UFF, a full periodic-table force-field for molecular mechanics and molecular-dynamics simulations[J].JournaloftheAmericanChemicalSociety, 1992, 114(25): 10024-10035. [27] Rappé A K, Goddard III W A. Charge equilibration for molecular-dynamics simulations[J].JournalofPhysicalChemistry, 1991, 95(8): 3358-3363. [28] Fukui K. The path of chemical-reactions-The IRC approach[J].AccountsofChemicalResearch, 1984, 14(12): 363-368. [29] Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian 09[CP]. Gaussian, Inc., Wallingford CT, 2009. [30] Megyes T, Bálint S, Grósz T, et al. Structure of liquid nitromethane: comparison of simulation and diffraction studies[J].JournalofChemicalPhysics, 2007, 126(16): 164507. [31] Wang L X, Yi C H, Zou, H T, et al. Rearrangement and thermal decomposition of nitromethane confined inside an armchair (5, 5) single-walled carbon nanotube[J].ChemicalPhysics, 2010, 367(2-3): 120-126. [32] Nguyen M T, Le H T, Hajgató B, et al. Nitromethane-methyl nitrite rearrangement: a persistent discrepancy between theory and experiment[J].JournalofPhysicalChemistryA, 2003, 107(21): 4286-4291. [33] Hu W F, He T J, Chen D M, et al. Theoretical study of the CH3NO2unimolecular decomposition potential energy surface[J].JournalofPhysicalChemistryA, 2002, 106(32): 7294-7303. [34] Sabourin J L, Dabbs D M, Yetter R A, et al. Functionalized graphene sheet colloids for enhanced fuel/propellant combustion[J].ACSNano, 2009, 3(12): 3945-3954.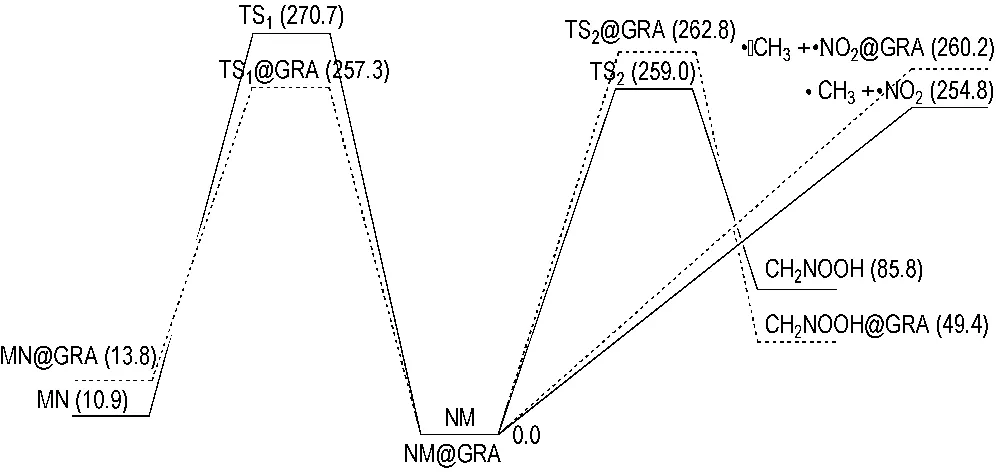
4 结 论
猜你喜欢
北京航空航天大学学报(2022年5期)2022-06-06石油化工自动化(2020年1期)2020-03-05青岛大学学报(工程技术版)(2019年2期)2019-09-10通信技术(2019年8期)2019-09-03国际呼吸杂志(2019年4期)2019-03-12电脑知识与技术(2018年3期)2018-03-21哈尔滨理工大学学报(2017年1期)2017-04-08科技视界(2016年24期)2016-10-11枣庄学院学报(2015年5期)2016-01-09中学化学(2015年8期)2015-12-29
