
几种芳烃加氢反应的热力学分析
2015-06-27 05:54胡意文达志坚王子军
石油学报(石油加工) 2015年1期
胡意文,达志坚,王子军
(中国石化 石油化工科学研究院,北京 100083)
几种芳烃加氢反应的热力学分析
胡意文,达志坚,王子军
(中国石化 石油化工科学研究院,北京 100083)
采用Benson基团贡献法计算得到萘、菲及芘加氢反应网络中各步反应在一定温度范围内的平衡常数,并系统分析了氢压、温度和物质结构对芳烃加氢平衡时的转化率、浓度分布和氢增量的影响。结果表明,加氢反应在低温下具有更好的热力学选择性,且氢压越高、温度越低时,那些芳环越少、环烷环越多及非取代芳香碳越多的芳烃具有越高的加氢平衡转化率。芳烃原料和其全加氢产物分别在高温低压、低温高压时热力学稳定,而部分氢化产物的热力学稳定区则位于两者之间,且对反应条件敏感。在芳烃各加氢产物中,全加氢产物具有较高的热力学选择性,部分氢化产物的热力学选择性较差。受热力学的限制,通过加氢来提高芳烃氢含量的效果有限。
芳烃加氢;热力学;Benson基团贡献法;平衡常数;平衡转化率;平衡浓度分布;氢含量
重质原油的高效轻质化是石油炼制行业亟待解决的问题,其关键是芳烃的轻质化,而芳烃加氢是实现该目的最为可行的方法之一。因此,研究芳烃的加氢转化意义重大[1]。
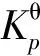
1 芳烃加氢反应和的计算
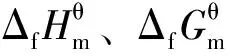
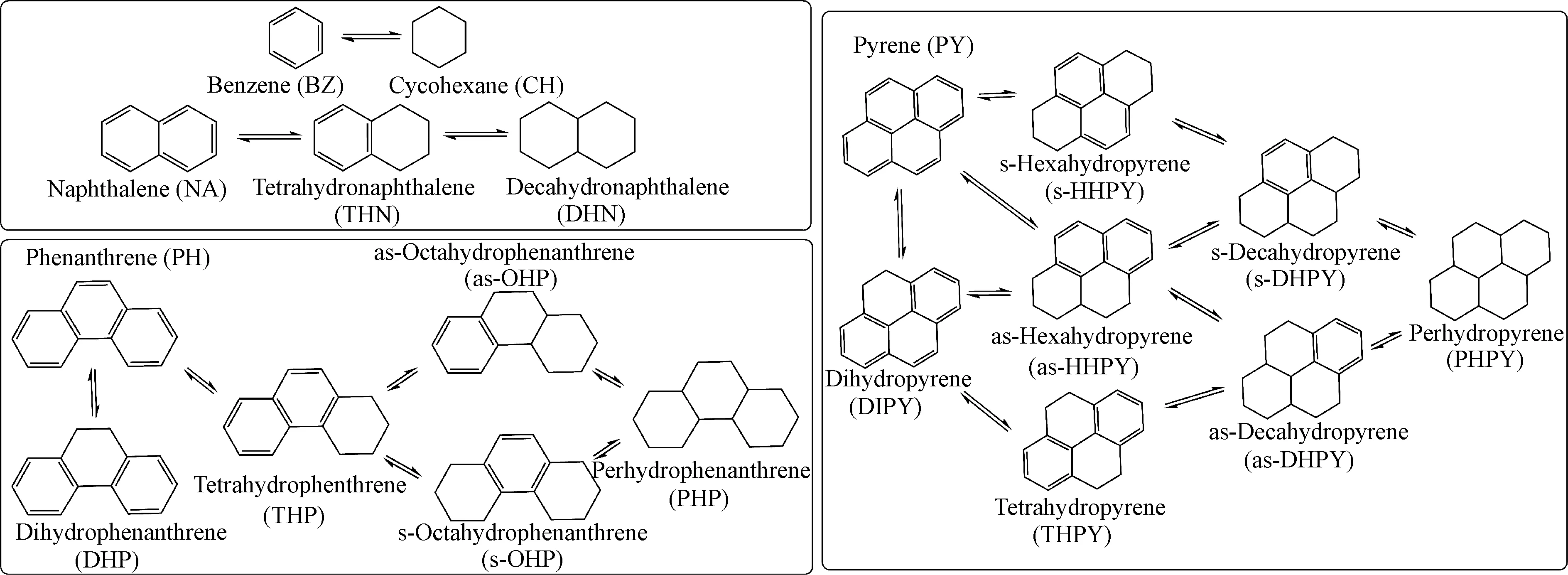
图1 几种芳烃的加氢反应网络[6,9-10]
表1 一些化合物的热力学数据
Table 1 Thermodynamic data of some compounds
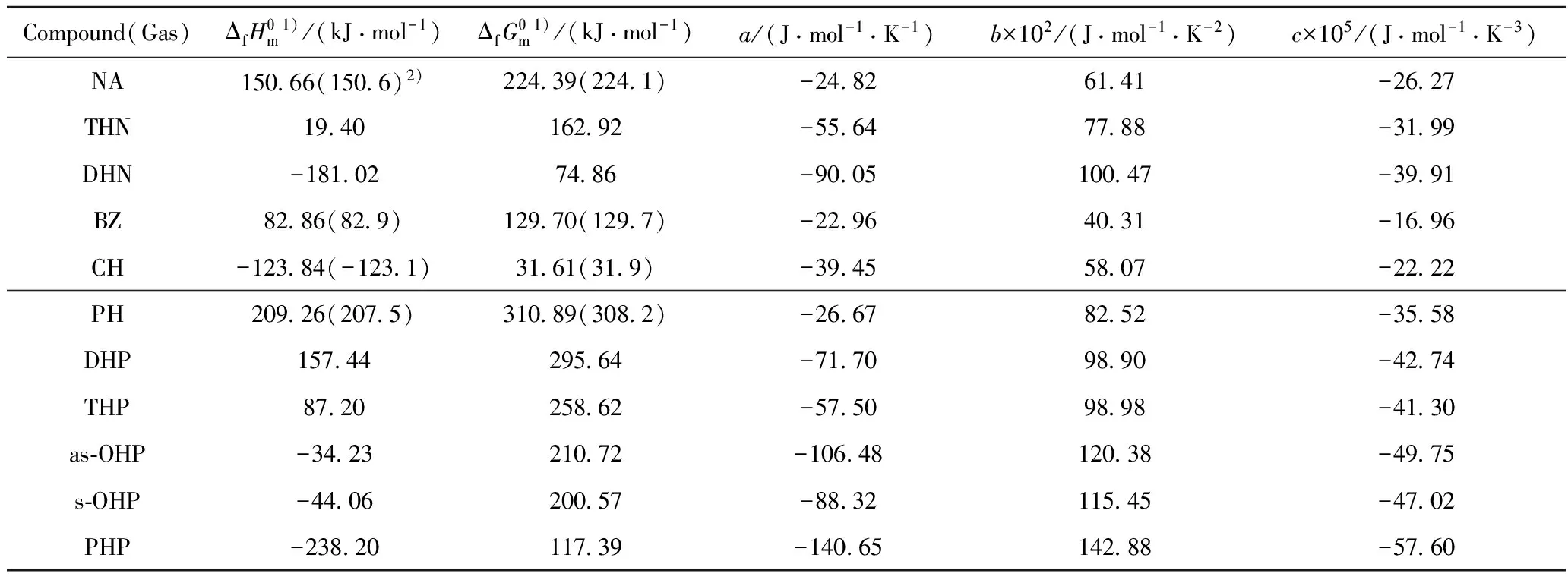
Compound(Gas)ΔfHθm1)/(kJ·mol-1)ΔfGθm1)/(kJ·mol-1)a/(J·mol-1·K-1)b×102/(J·mol-1·K-2)c×105/(J·mol-1·K-3)NA150 66(150 6)2)224 39(224 1)-24 8261 41-26 27THN19 40162 92-55 6477 88-31 99DHN-181 0274 86-90 05100 47-39 91BZ82 86(82 9)129 70(129 7)-22 9640 31-16 96CH-123 84(-123 1)31 61(31 9)-39 4558 07-22 22PH209 26(207 5)310 89(308 2)-26 6782 52-35 58DHP157 44295 64-71 7098 90-42 74THP87 20258 62-57 5098 98-41 30as⁃OHP-34 23210 72-106 48120 38-49 75s⁃OHP-44 06200 57-88 32115 45-47 02PHP-238 20117 39-140 65142 88-57 60
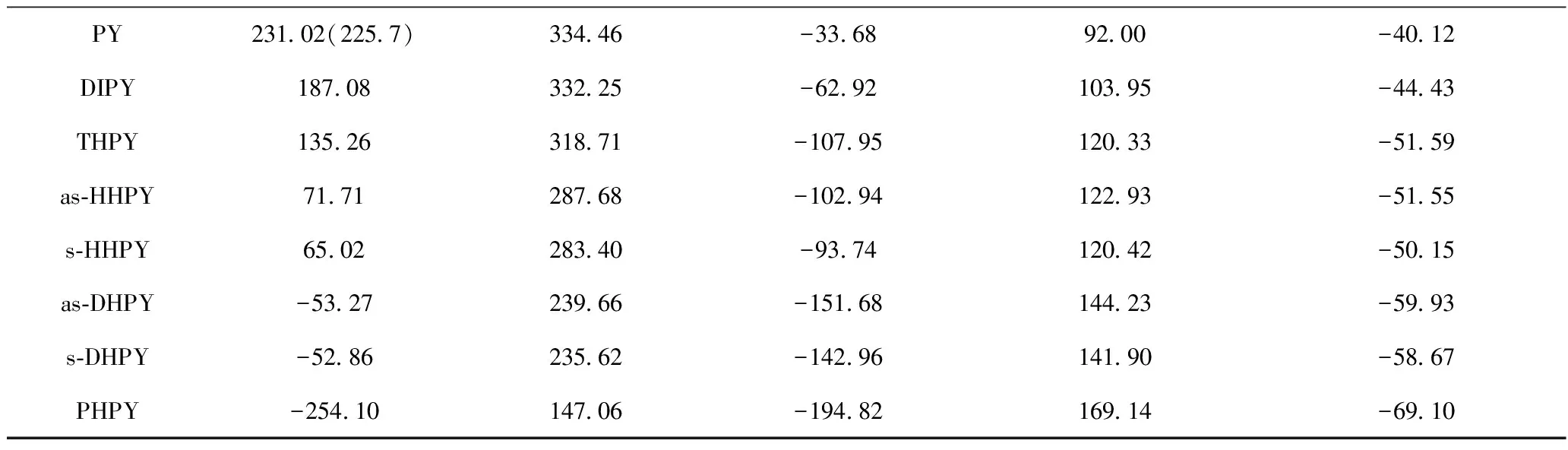
续表1
1) Obtained at 298.15 K; 2) The data in the bracket were experimental values adopted from CRC handbook of chemistry and physics (90th Edition)
需要说明的是,Benson法一般只能估算某物质在气态时的热力学数据,因此接下来的各类计算均基于最理想的情况,即假定所涉物种均处于气态。
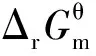
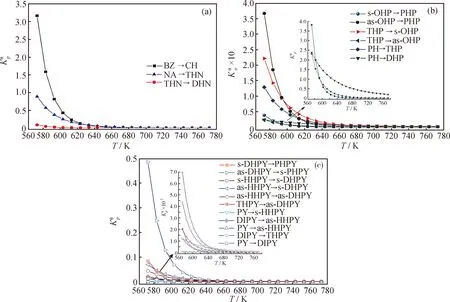
图2 苯、萘、四氢萘、菲系列及芘系列物种加氢反应的标准平衡常数)随温度(T)的变化
由图2可知:(1)所有反应的标准平衡常数均随温度的升高而降低,表明加氢反应为放热反应,低温有利于加氢反应平衡右移。(2)标准平衡常数随温度的变化曲线大多呈“L”型,可将其大致分为低温急减区、中温缓减区和高温微减区3个区域。573~613 K属于低温急减区,标准平衡常数随着温度升高迅速减小;613~673 K属于中温缓减区,标准平衡常数随着温度升高缓慢减小;673~773 K属于高温微减区,标准平衡常数随着温度升高略微减小。在低温区,热力学对温度敏感,升温需谨慎;中温区,温度可在一定范围内调节,热力学上的影响不大;高温区,大幅升高温度对热力学的影响也较小。(3)NA、PH和PY系列物种的标准平衡常数在低温下相差较大,而在高温下相差很小,这表明加氢反应在低温下具有更好的热力学选择性。(4)总的来讲,随着芳环数的增加,加氢反应的标准平衡常数减小,每增加1个芳环,平衡常数减小至原来的10%~20%,而THN、THP和DIPY的计算数据表明,环烷环的存在能减缓甚至逆转这种减小趋势,即芳烃上并合有环烷环对其加氢反应热力学是有利的。(5)不同物种的变化曲线往往会重合、平行或交叉,表明物质结构和温度对加氢反应热力学具有十分复杂的影响。
2 不同芳烃的加氢平衡转化率
为了考察氢压、温度和物质结构对芳烃加氢平衡转化率的影响,将图1中各步反应单独处理,即假定其仅存1种反应物、1种生成物和H2;H2接近理想气体,认为其活度系数为1;并近似认为芳烃和其加氢产物的活度系数相同。则各反应物的平衡转化率如式(1)所示。
(1)
从已计算得到的各芳烃加氢反应在不同温度下的标准平衡常数,再通过式(1)就可求得各反应物在不同条件(温度、氢压)下的平衡转化率。
2.1 氢压对芳烃加氢平衡转化率的影响
以PY系列物种为例,计算在673 K时氢压对其加氢平衡转化率的影响,结果如图3所示。由图3可知,芳烃的加氢平衡转化率均随氢压升高而增大,但增加趋势有所不同,可大致分为类线性型(如DIPY加氢为THPY)、内凹型(如PY加氢为s-HHPY)、外凸型(如DIPY加氢成as-HHPY)3类。具有类线性型曲线的加氢反应的转化率随着氢压增大近似线性地增加;具有内凹型曲线的加氢反应的转化率在氢压较低时随氢压变化的幅度较小,当氢压增至拐点后,转化率随着氢压增大开始显著增大;具有外凸型曲线的加氢反应的转化率在低氢压时增幅显著,越过拐点后再增加氢压,转化率提高有限。显然,具有外凸型曲线的加氢反应在较低氢压时就能获得很高的转化率,而具有前两类曲线的加氢反应则需较高的氢压才能获得较好的转化率。
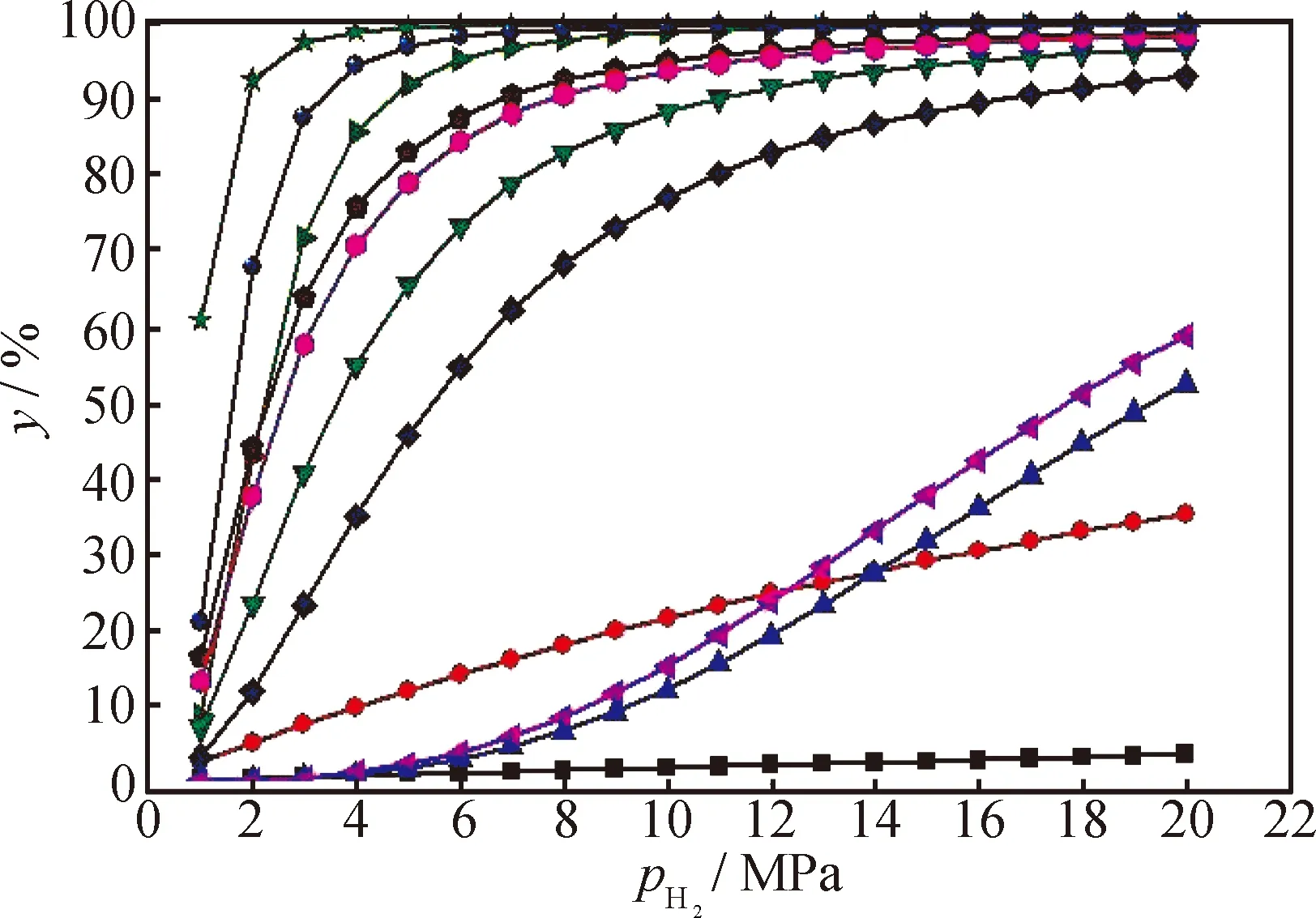
图3 芘系列物种加氢平衡转化率(y)随氢压(pH2)的变化
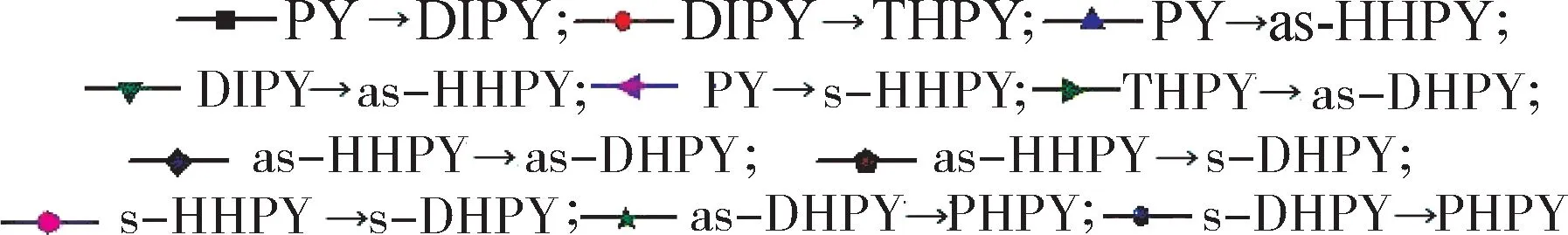
2.2 温度对芳烃加氢平衡转化率的影响
以PY系列物种为例,计算在pH2=1 MPa时温度对其加氢平衡转化率的影响,结果如图4所示。由图4可知,各芳烃的加氢平衡转化率随温度的变化曲线与其标准平衡常数的变化曲线有相似之处,如随温度升高而减小、在不同温度区域减小幅度不同、低温下“分级”以及曲线间交叉平行重合等。但pH2的引入也使其呈现出一些新的特点。(1)平衡转化率随温度的变化曲线不仅存在“L”型,还出现“S”型。“L”型的“急减区”仍在低温区,但减小的幅度大为缓和;而“S”型的“急减区”则从低温区移至中温甚至高温区,这些变化表明了pH2对平衡右移的促进作用。(2)较大的温度范围内,具备“S”型曲线的物种的加氢平衡转化率普遍比“L”型的要高。(3)具备“S”型曲线的物种的热力学敏感区处于中温区,与具备“L”型曲线的物种处于低温区有所不同,因此在选择合适反应温度时要区别对待。
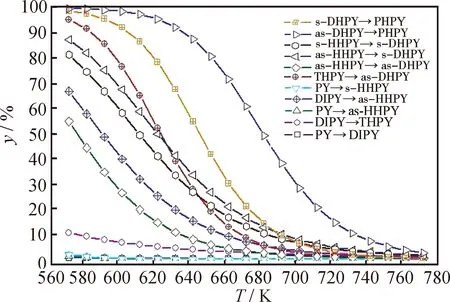
图4 芘系列物种的加氢平衡转化率(y)随温度(T)的变化
2.3 物质结构对芳烃加氢平衡转化率的影响
为了研究物质结构对芳烃加氢转化的影响,将所计算的芳烃物种从芳环数、环烷环数、非取代芳香碳个数3个方面进行细分,当比较芳环数相同的物种时,则考虑环烷环数及芳环之间、芳环与环烷环之间联结位置(即非取代芳香碳个数)对热力学平衡的影响;当比较环烷环数相同的物种时,则考虑芳环数及非取代芳香碳个数对热力学平衡的影响。
2.3.1 芳环数相同
不同芳环数的几组反应物的平衡转化率随温度的变化如图5所示。由图5可知,(1)对于相同芳环数的芳烃,其并合的环烷环数越多、非取代芳香碳越多,其加氢反应的平衡转化率就越大。在环烷环和非取代芳香碳两因素中,又往往是后者对平衡右移的促进作用更大。(2)对于反应物相同、产物不同的加氢反应,其平衡转化率主要受产物的非取代芳香碳数的影响,产物中非取代芳香碳越多,越不利于加氢平衡右移。(3)反应耗氢量越大,氢压对反应平衡的促进作用就越明显,另外分子的对称性、环烷环张力也对加氢热力学有影响。
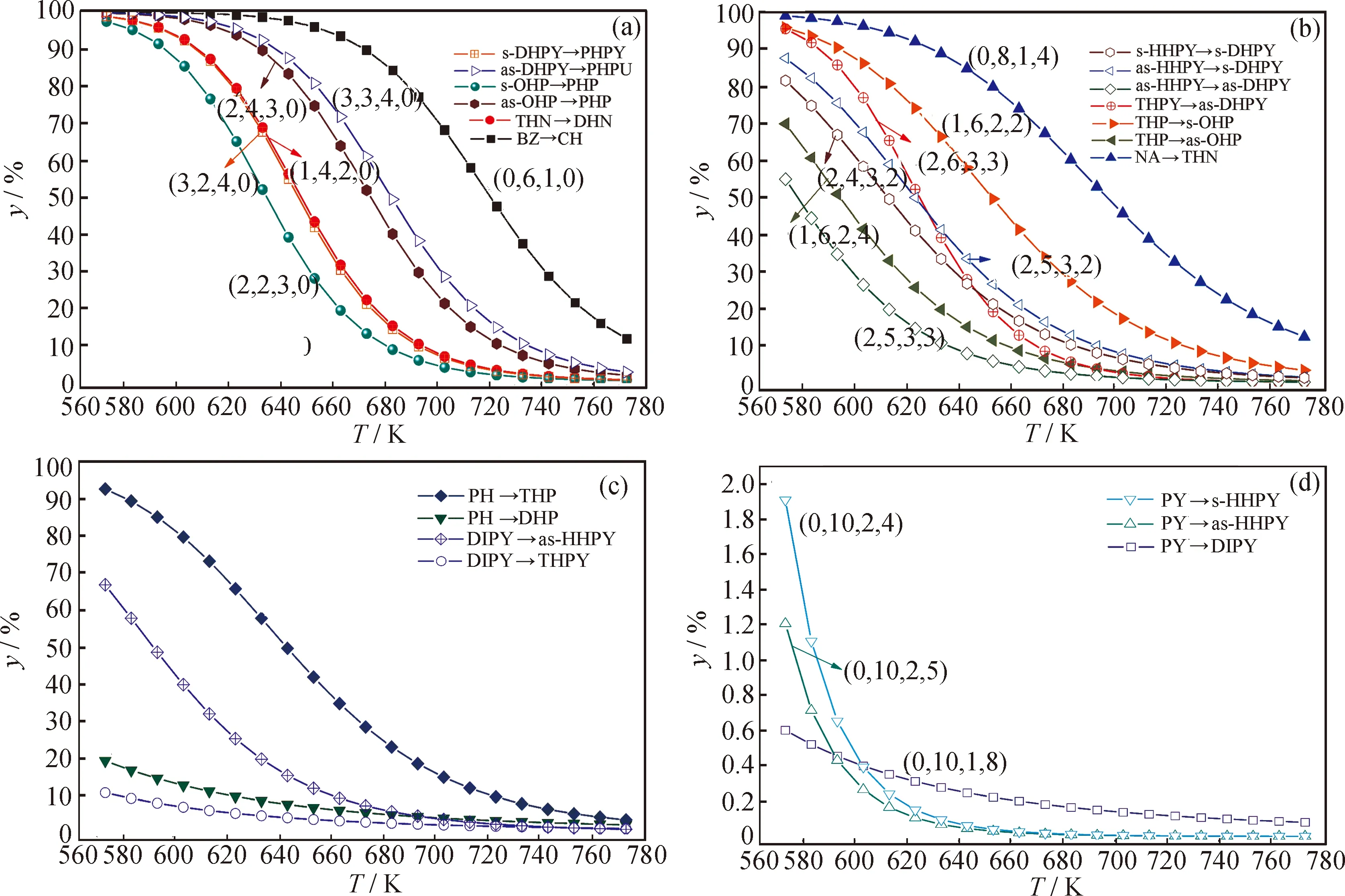
图5 不同芳环数的几组芳烃加氢反应平衡转化率(y)随温度(T)的变化
2.3.2 环烷环数相同
图6为不同环烷环数的几组反应物加氢反应平衡转化率随温度的变化。由图6可知,反应物中芳环数越多,其平衡转化率就越小;非取代芳香碳数越多,其平衡转化率越大。一般而言,当反应物相同时,产物的非取代芳香碳数越多,反应物的平衡转化率越小。
物质结构对芳烃加氢平衡的影响较为复杂。一般而言,芳环数的影响最为显著,随着芳环数增加,平衡转化率大幅下降,而环烷环则有利于减缓该下降趋势。环之间的并合方式在此用非取代芳香碳数来标识,非取代芳香碳数较少的芳烃容易生成却不容易被进一步转化,表现出较高的热力学稳定性,s-OHP、s-HHPY、s-DHPY俱是如此。
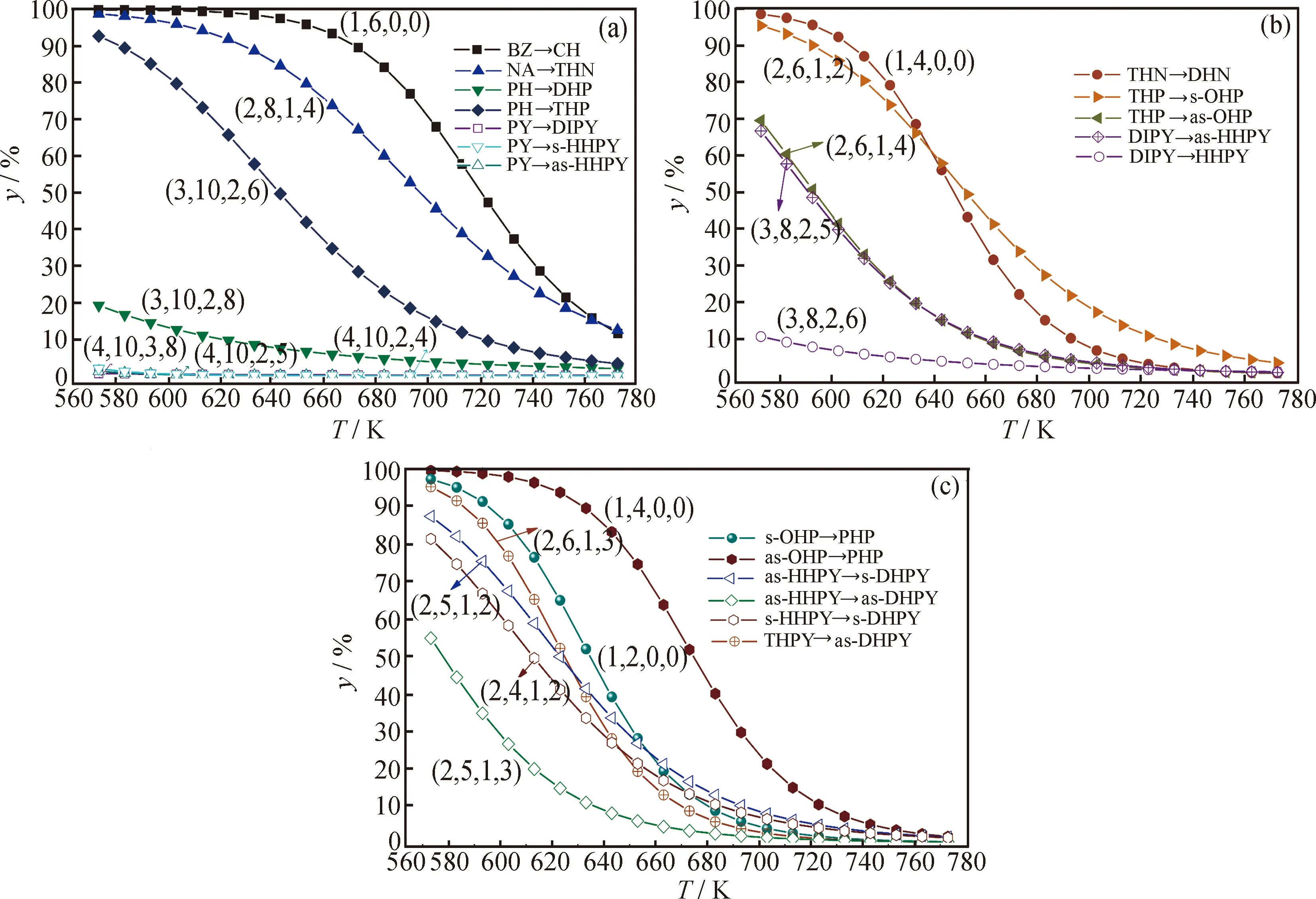
图6 不同环烷环数的几组芳烃加氢反应平衡转化率(y)随温度(T)的变化
3 芳烃连续加氢反应的平衡浓度分布
在上述单步加氢反应热力学分析的基础上,以连续反应体系为分析对象,计算各物质的平衡态浓度,并获得其浓度分布随温度的变化曲线。
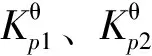
(2)
(3)
xDHN+xTHN+xNA=1
(4)
由式(2)~(4)及一定温度下的标准平衡常数,就可计算出该温度下NA连续加氢体系中各物种的平衡浓度。PH和PY连续加氢体系中各物种平衡浓度的计算方法与此类似,不再赘述。
NA、PH和PY各物种的平衡浓度在一定氢压和温度范围内的变化趋势分别如图7~图9所示,为清楚直观,忽略了各图的z坐标尺度,故三维峰高并不反映各物种的真实浓度大小。
从图7~9可知,各物种的平衡浓度在一定温度和氢压组成的区域内都有其特征。可将分布图分为“瀑布”型和“山脉”型两类。NA、PH、PY和DHN、PHP、PHPY属于“瀑布”型,它们的分布图中有高浓度区(构成“瀑布”的帘顶)、浓度急剧减小区(构成“瀑布”的帘面)和低浓度区(构成“瀑布”的帘底)3个区域。原料的“帘顶”处于高温低压区,“帘底”处于低温高压区,全加氢产物则恰好相反,这说明低温、高氢压有利于芳烃的加氢转化。部分加氢产物的三维分布图均为“山脉”型,即在一定温度-氢压区域内具有浓度峰值。图10为各物种加氢反应热力学稳定区示意图。图10中A区域为高温低压区,C区域为低温高压区,B区域为介于二者之间的区域。
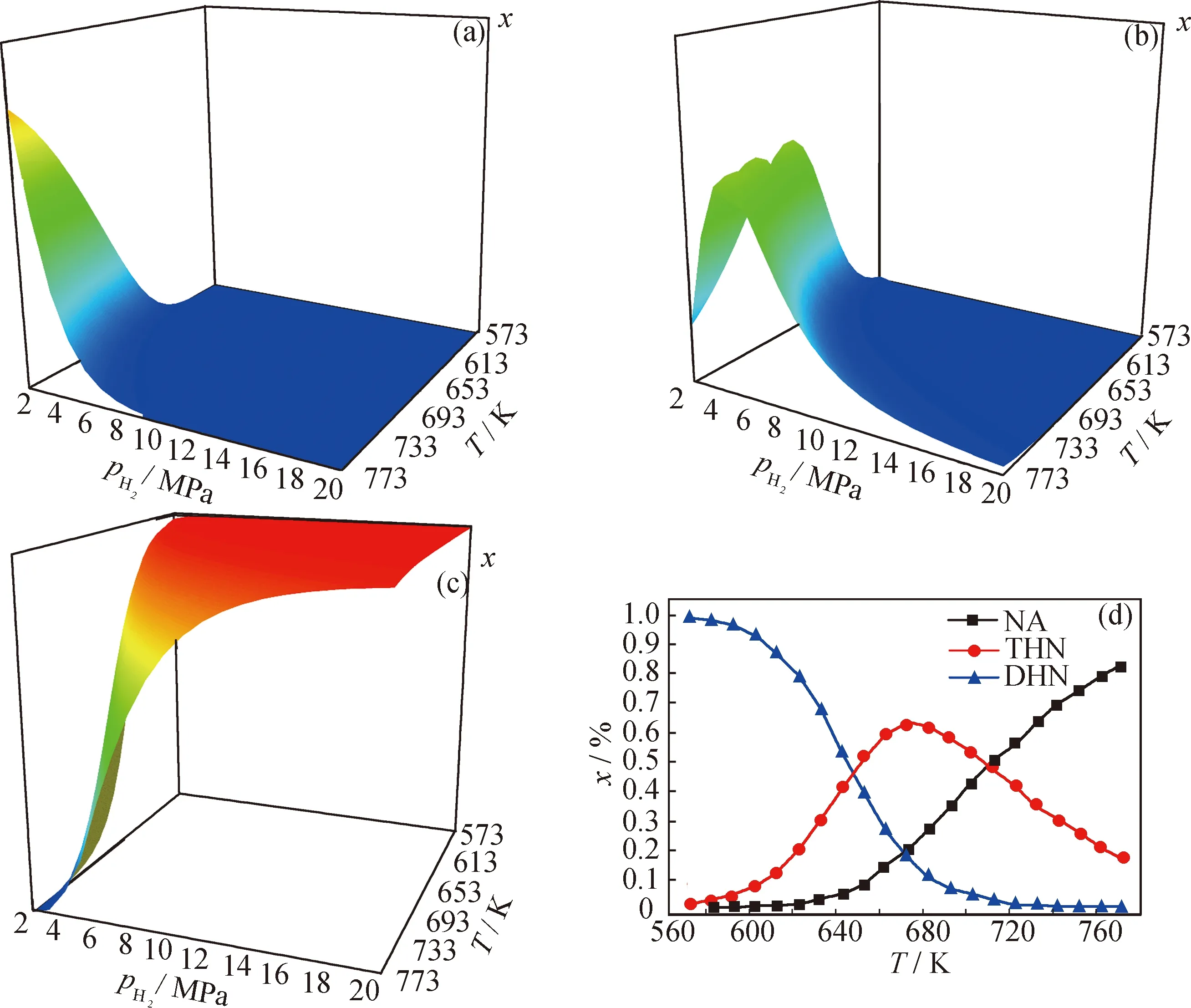
图7 萘连续加氢体系中各物种平衡浓度(x)随氢压(pH2)和温度(T)的变化
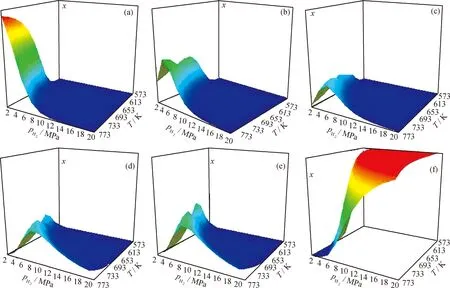
图8 菲连续加氢体系中各物种平衡浓度(x)随氢压(pH2)和温度(T)的变化

图9 芘加氢体系中各物种平衡浓度(x)随氢压(pH2)和温度(T)的变化
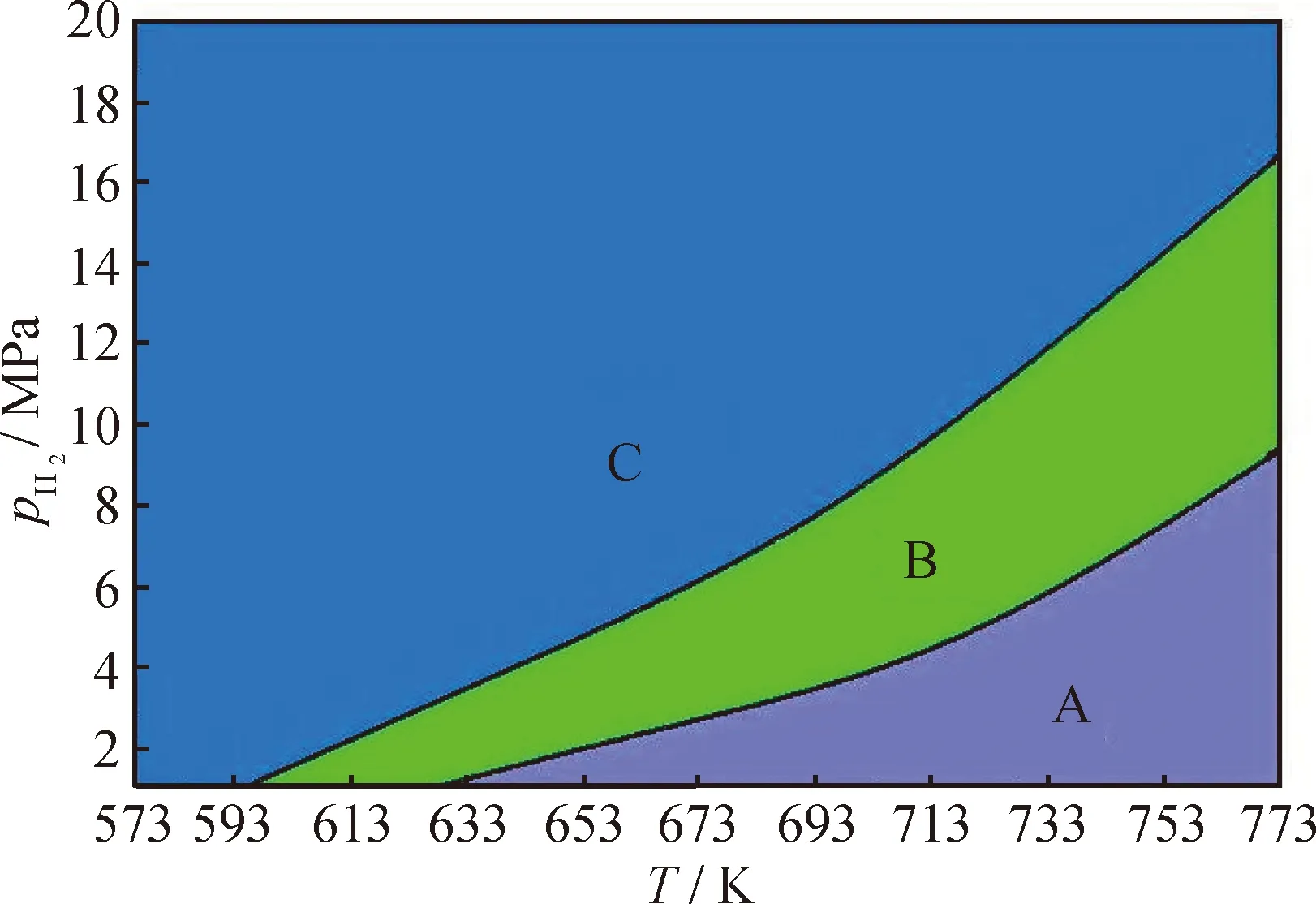
图10 各物种加氢反应热力学稳定区示意图
三维分布图能很好地反映加氢体系中各物种的热力学稳定性。显然,物种的平衡浓度越大,其热力学稳定性越好,据此可判断,原料的热力学稳定区域位于高温低压区(图10中A区域),其芳环数越多,该区域就越向外扩大,说明环数越多的芳烃越难被加氢转化。全加氢产物的热力学稳定区则在低温高压区(图10中C区域),其环数越多,该区域越向内缩小,说明环数越多的全氢产物越难被加氢生成。部分氢化产物的热力学稳定区则介于两者之间(图10中B区域),且氢化程度越高,其热力学稳定区越靠近C区域。与原料和全加氢产物相比,部分氢化产物的热力学稳定区都较小,且相互重叠,显示出其在热力学上稳定性差和性质相近的特点。
从浓度分布上看,A区以原料为主,C区以全加氢产物为主,而B区则对应着原料和全加氢产物的“瀑布帘面”,可看作是前者向后者转化的过渡区,该区含有原料和氢化程度不同的各种产物,且其不同子区域中有着不同的浓度分布。有些子区域内十氢、八氢等氢化程度高的产物占优,而有些子区域内则二氢、四氢等氢化程度低的产物占优。这表明,当选择B区的反应条件时,芳烃加氢对温度、氢压有着较强的热力学敏感性,稍微改变反应条件,其产物分布就会发生很大的变化。反观A和C区,即使大范围的改变反应条件,产物分布几乎不变,表现出很强的热力学惰性。
取各物种三维分布图在某氢压的截面就可得到其二维分布图。由于篇幅所限,笔者只给出了氢压为1 MPa时的二维图(见图7(d)、图11)。在二维图的统一坐标尺度下,能很好地比较各物种的浓度大小及其随温度的变化趋势。
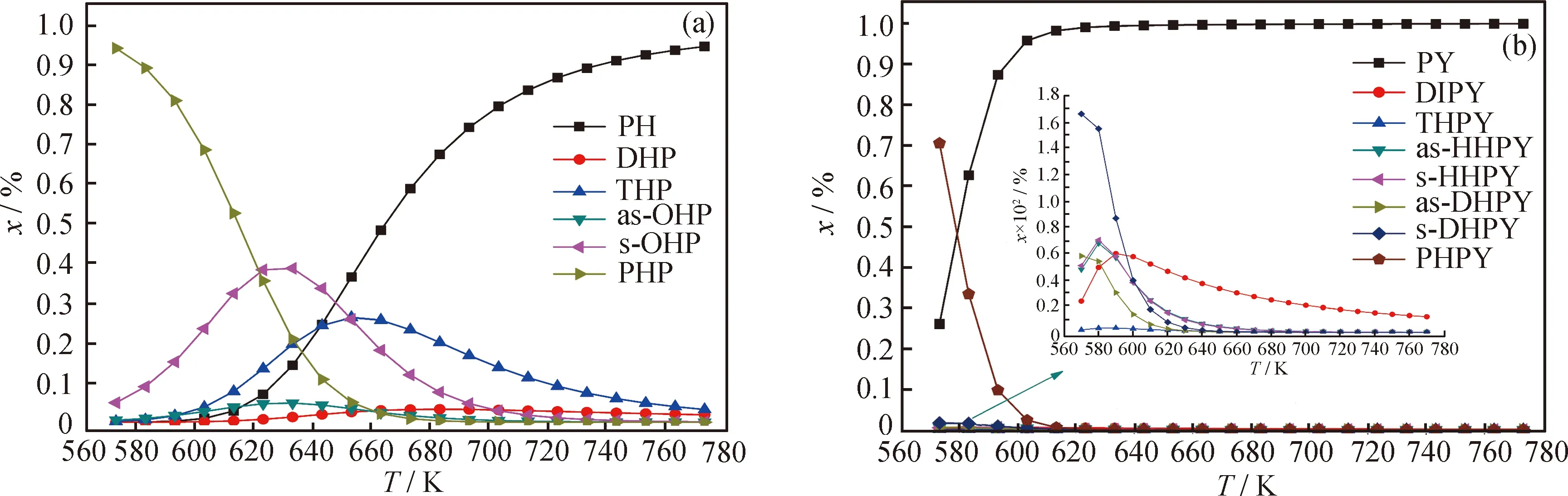
图11 菲、芘各物种加氢反应平衡浓度(x)随温度(T)的变化
NA、PH和PY的平衡浓度都随温度的升高而增大,随着芳环数增多,此增大趋势更为明显。如NA和PH都呈“S”型增大,直到773 K时,仍有约20%的NA加氢产物和0.5%的PH加氢产物;PY则呈“L”型增大,在623 K时,反应体系中仅有痕量的加氢产物;全加氢产物则与之相反。中间加氢产物平衡浓度大多随温度呈“正态分布”,具有浓度峰值;随着芳环数的增加,“正态分布”曲线逐渐变“矮”,且峰值向低温方向移动。因此,随着温度升高,可大致将中间加氢产物平衡浓度分布分为三段,即低温、高转化、全加氢产物段;中温、中等转化、混合加氢产物段;高温、低转化、原料段。芳烃不同,其各段的温度范围和物质组成会有所差异。
不难发现,在各芳烃加氢产物中,只有全氢产物具有较高的热力学选择性,也就是说,若反应条件合适,可通过延长反应时间来获得较纯净的全加氢产物,而部分氢化产物则很难经此获得;又,平衡浓度从大到小的顺序依次为s-OHP、as-OHP、s-DHPY、as-DHPY,THPY浓度极低,说明端环加氢在热力学上的选择性高,而内环则非常低。
4 芳烃连续加氢反应的氢增量
平衡态的氢元素增加量(ΔwH,以原料中氢元素的质量分数为基准)可直观地衡量芳烃加氢转化的难易程度。因此在计算平衡浓度的基础上,进一步考察氢增量随氢压和温度的变化。
图12为各芳烃加氢平衡时氢元素增加量随氢压和温度的变化。由图12可知,在相同反应条件下,NA的氢增量最大,而PY最小。也就是说,要获得相同的氢增量,PY需要更高的氢压或更低的温度,更低的温度则意味着更长的反应时间。因此,芳烃的环数越多,其加氢的难度就越大。如在1 MPa氢压下,想通过加氢使原料的氢含量增加2.0%,NA的反应温度不超过693 K,PH需低于653 K的反应温度,而PY仅能在588 K以下进行才可。
芳烃加氢是重油轻质化的关键步骤,其目标是在较低的氢压下快速增加氢含量,但芳烃加氢的热力学特性表明,实现该目标并不容易,芳环越多,难度就越大。可以预见,由于受热力学的限制,大片状稠环芳烃即使在低温、高氢压下长时间反应,也仅可获得有限的氢增量。因此,引入裂化功能,在加氢过程中减少芳环数或许是芳烃高效转化的有利方法。
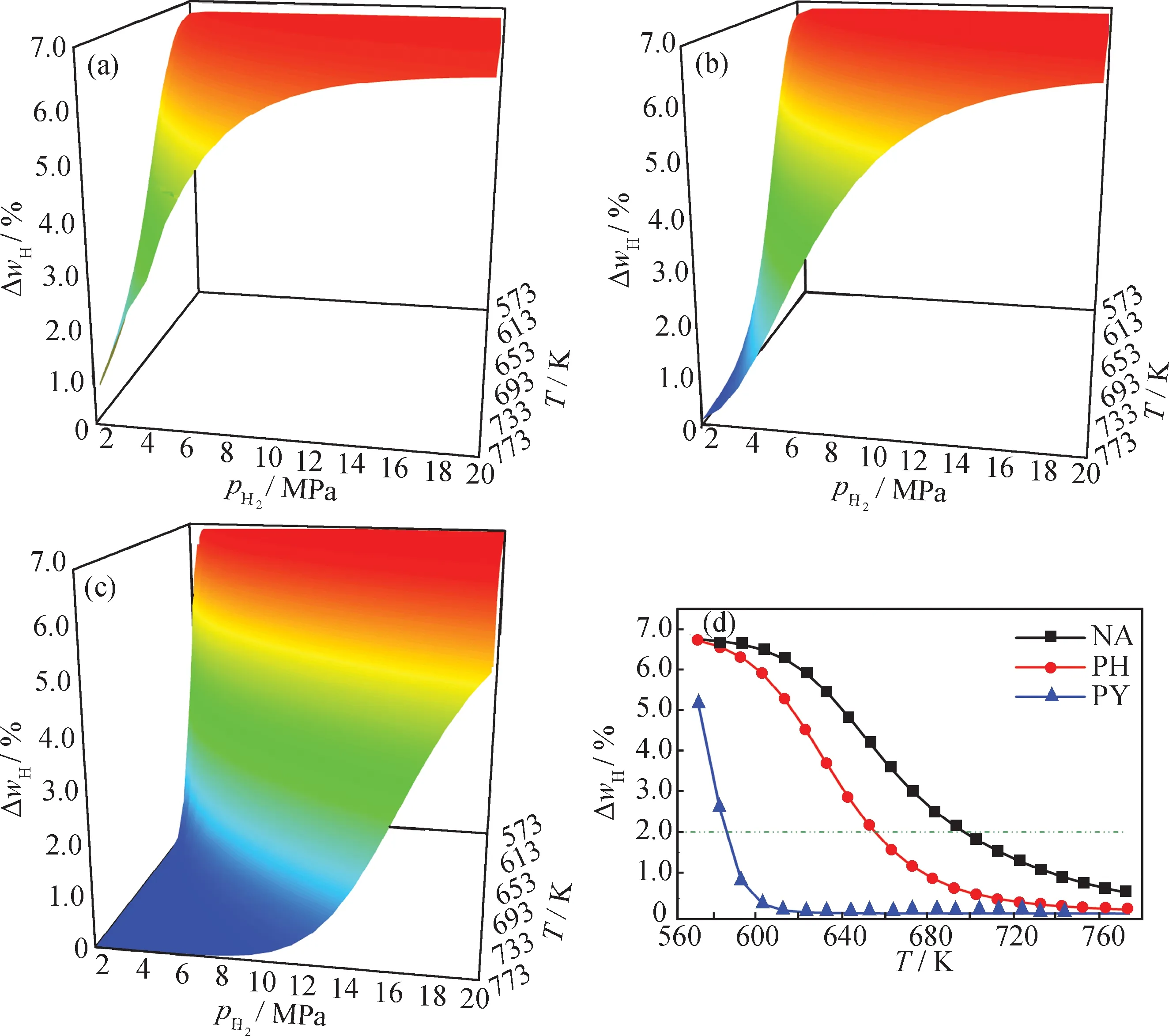
图12 各芳烃加氢平衡时氢增量(ΔwH)随氢压(pH2)和温度(T)的变化
5 结 论
(1)芳烃加氢反应为放热反应,平衡常数随温度的升高而减小,但在不同温度区,减小幅度相差很大,可大致分为低温急减区、中温缓减区和高温微减区。
(2)萘、菲和芘系列物种的标准平衡常数在低温下都出现分级,而高温下相差较小,说明加氢反应在低温下具有更好的热力学选择性。
(3)芳烃加氢的平衡转化率随氢压的增高而加大,随温度的升高而减小。芳环数、环烷环数及非取代芳香碳数综合影响着芳烃的加氢平衡;一般而言,芳烃的芳环越少、环烷环越多、非取代芳香碳越多,其加氢反应的平衡转化率就越大。
(4)分析芳烃连续加氢反应体系中各物种的平衡浓度随氢压和温度的变化情况后发现,原料的热力学稳定区位于高温低压处,环数越多,区域越大;全加氢产物的热力学稳定区位于低温高压处,环数越多,区域越小;部分氢化产物的热力学稳定区位于两者之间,但范围小,且相互重叠。
(5)原料和全加氢产物的热力学稳定区呈热力学惰性,大幅改变反应条件,各物种的浓度分布几乎不变;而部分氢化产物的热力学稳定区呈热力学敏感性,稍微改变反应条件,各物种的浓度分布就会发生很大的变化。
(6)全氢产物具有较高的热力学选择性,在合适的反应条件下,通过延长反应时间可获得较纯净的产品,而部分氢化产物则无法做到。
(7)受热力学的限制,通过加氢来提高芳烃氢含量的效果有限。
符号说明:
as-DHPY——as-十氢芘;
as-HHPY——as-六氢芘;
as-OHP——as-八氢菲;
BZ——苯;
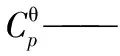
CH——环己烷;
DHP——二氢菲;
DHN——十氢菲;
DIPY——二氢芘;
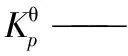
NA——萘;
pθ——标准压力,0.1 MPa;
pH2——氢气压力,MPa;
PH——菲;
PHP——全氢菲;
PY——芘;
PHPY——全氢芘;
R——理想气体常数,J/(mol·K);
s-DHPY——s-十氢芘;
s-HHPY——s-六氢芘;
s-OHP——s-八氢菲;
T——热力学温度,K;
THN——全氢芘;
THP——四氢菲;
THPY——四氢芘;
xi——i组分摩尔分数;
yi——i组分平衡转化率;
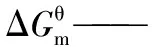
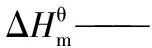
ΔwH——平衡态的氢元素增加量,以原料中氢元素的质量分数为基准,%。
[1] 聂红, 杨清河, 戴立顺, 等. 重油高效转化关键技术的开发及应用[J]. 石油炼制与化工, 2012, 43(1): 1-6. (NIE Hong, YANG Qinghe, DAI Lishun, et al. Development and commercial application of key technology for efficient conversion of heavy oil[J]. Petroleum Processing and Petrochemicals, 2012, 43(1): 1-6.)
[2] GIRGIS M J, GATES B C. Reactivities, reaction networks, and kinetics in high-pressure catalytic hydroprocessing[J]. Ind Eng Chem Res, 1991, 30(9): 2021-2058.
[3] 张全信, 刘希尧. 多环芳烃的加氢裂化[J]. 工业催化, 2001, 9(2): 10-16. (ZHANG Quanxin, LIU Xiyao. Hydrocracking of polycyclic aromatic hydrocarbons[J]. Industrial Catalysis, 2001, 9(2): 10-16.)
[4] RASMUSSEN J A. Polycyclic aromatic hydrocarbons: Trends for bonding hydrogen[J]. J Phys Chem A, 2013, 117: 4279-4285.
[5] BOUKHVALOV D W, FENG X, MULLEN K. First-principles modeling of the polycyclic aromatic hydrocarbons reduction[J]. J Phys Chem C, 2011, 115: 16001-16005.
[6] KORRE S C, KLEIN M T, QUANN R J. Polynuclear aromatic hydrocarbons hydrogenation 1 Experimental reaction pathways and kinetics[J]. Ind Eng Chem Res, 1995, 34(1): 101-117.
[7] CONTRERAS M S, de BRUIN T, MOUGIN P, et al. Thermochemistry of 1-methylnaphthalene hydro- conversion: Comparison of group contribution and ab-initio models[J]. Energy & Fuels, 2013, 27(9): 5475-5782.
[8] POLING B E, PRAUSNITZ J M, O’CONNELL J P著.赵红玲, 王凤坤, 陈圣坤等译. 气液物性估算手册[M]. 第五版. 北京:化学工业出版社,2006:60-85.
[9] LAPINAS A T, KLEIN M T, MACRIS A, et al. Catalytic hydrogenation and hydrocracking of fluoranthene: Reaction pathways and kinetics[J]. Ind Eng Chem Res, 1987, 26(5): 1026-1033.
[10] LAPINAS A T, KLEIN M T, GATES B C, et al. Catalytic hydrogenation and hydrocracking of fluorene: Reaction pathways, kinetics, and mechanisms[J]. Ind Eng Chem Res, 1991, 30(1): 42-50.
Thermodynamic Analysis on the Hydrogenation Reaction of Several Aromatic Hydrocarbons
HU Yiwen,DA Zhijian,WANG Zijun
(ResearchInstituteofPetroleumProcessing,SINOPEC,Beijing100083,China)
The equilibrium constants in a certain temperature range of every step conversion in the hydrogenation reaction networks of naphthalene, phenanthrene and pyrene were calculated with Benson group-contribution method. And the effects of hydrogen pressure, temperature and material structure to the equilibrium conversion, equilibrium concentration distribution and hydrogen-content increment of aromatic hydrogenation were systematically analyzed. The results showed that a significant thermodynamic selectivity of hydrogenation reaction seemed to be achieved only in low temperature zone, and when those aromatic hydrocarbons with less aromatic rings, more naphthenic rings and more non-substituted aromatic carbons were hydrogenated at higher hydrogen pressure and lower temperature, a higher equilibrium conversion could be obtained. The thermodynamic stabilization of reactant and perhydro-products was preserved at high temperature and low hydrogen pressure conditions, or low temperature and high hydrogen pressure conditions, respectively, while the thermodynamic stabilization of partial hydrogenation products was preserved at the middle conditions, which exhibited strong thermodynamic sensitivity to the reaction conditions. Among the hydrogenation products of aromatic hydrocarbons, perhydro-products held high thermodynamic selectivity compared with partial hydrogenation products. Also, the effect of hydrogen-content increase in aromatics by hydrogenation reaction was limited because of the thermodynamic restriction.
aromatic hydrogenation; thermodynamics; Benson group-contribution method; equilibrium constant; equilibrium conversion; equilibrium concentration distribution; hydrogen content
2014-08-14
国家重点基础研究发展计划“973”项目(2012CB224801)基金资助
胡意文,男,博士研究生,从事芳烃转化方面的研究;Tel:010-82368362;E-mail:huyiwen.ripp@sinopec.com
1001-8719(2015)01-0007-11
O625.15;O642.4
A
10.3969/j.issn.1001-8719.2015.01.002
猜你喜欢
机械工业标准化与质量(2022年6期)2022-08-12
科学家(2021年24期)2021-04-25
中国调味品(2017年2期)2017-03-20
电子制作(2016年19期)2016-08-24
制冷技术(2016年4期)2016-08-21
橡胶工业(2015年9期)2015-08-29
橡胶工业(2015年6期)2015-07-29
橡胶工业(2015年4期)2015-07-29
中学化学(2015年2期)2015-06-05
山西大同大学学报(自然科学版)(2015年1期)2015-01-22
