
猪伪狂犬病毒gB和gC蛋白B细胞表位编码序列的融合表达及活性测定
2016-02-06 07:18邓瑞广张改平李学伍
河南农业科学 2016年1期
张 冲,刘 芳,赵 东,邓瑞广,张改平,3,李学伍*
(1.河南科技大学 动物科学技术学院,河南 洛阳 471003; 2.河南省农业科学院 动物免疫学重点实验室,河南 郑州 450002; 3.河南农业大学 牧医工程学院,河南 郑州 450002)
猪伪狂犬病毒gB和gC蛋白B细胞表位编码序列的融合表达及活性测定
张 冲1,2,刘 芳1,赵 东2,邓瑞广2,张改平2,3,李学伍2*
(1.河南科技大学 动物科学技术学院,河南 洛阳 471003; 2.河南省农业科学院 动物免疫学重点实验室,河南 郑州 450002; 3.河南农业大学 牧医工程学院,河南 郑州 450002)
依据猪伪狂犬病毒gB和gC蛋白的B细胞表位编码序列,分别设计gB和gC蛋白B细胞表位编码序列的融合扩增引物,利用融合PCR技术,将gB和gC蛋白B细胞表位编码序列有机地融合为gB-C表位编码序列。将融合基因片段插入到原核表达载体pET28a,并转化大肠杆菌BL21(DE3),1.0 mmol/L异丙基硫代半乳糖苷(IPTG)诱导表达重组蛋白。SDS-PAGE结果显示,融合蛋白gB-C能够高效表达,重组蛋白的分子质量约为38 ku;经Western blot鉴定分析,小鼠抗His标签单克隆抗体和PRV阳性血清均能识别表达的重组蛋白。表明成功表达了融合蛋白gB-C,且表达的重组蛋白具有较好的免疫学活性。
猪伪狂犬病毒; gB蛋白; gC蛋白; B细胞表位; 融合表达
猪伪狂犬病是由猪伪狂犬病毒(PRV)引起,造成母猪严重繁殖障碍以及仔猪高死亡率的传染病。PRV感染导致机体免疫抑制,引起其他病原如猪圆环病毒、猪链球菌和猪肺炎支原体等的继发性感染和混合感染,是危害养猪业最严重的病毒性疾病之一[1-2]。PRV 基因组为线状双链 DNA 分子,大小约为 150 kb,G+C 含量高达 73%,分子质量约 9.5×103ku,属线状双链DNA 病毒。糖蛋白 gB(gⅡ)、gC(gⅢ)均被定位于UL区。到目前为止,已证实 gB对病毒体内增殖是非必需的,而 gC为病毒复制所必需[3]。在疱疹病毒中,gB蛋白既是良好的保护性抗原又是可靠的免疫检测抗原。gB蛋白由gBa、gBb和gBc 3个糖蛋白通过二硫键共价连结而成。gBb、gBc由gBa剪切而来;gB蛋白通常包含2个gBb和2个gBc分子。gBc、gBb存在多个结构表位和线性表位[4-5]。gB蛋白诱导产生的抗体能中和病毒阻止感染传代细胞,gB蛋白免疫的小鼠能够抵抗PRV的攻击免受病毒的感染。gC蛋白也是PRV的重要功能蛋白质之一,广泛存在于PRV囊膜表面[6],gC蛋白能够与宿主细胞表面的硫酸乙酰肝素蛋白(HS)结合,从而启动病毒对细胞的侵入,病毒囊膜与细胞膜融合始于gC蛋白与HS的结合[7]。gC蛋白诱导产生的中和抗体主要识别HS结合区域内的B细胞表位[8]。gC蛋白单抗能够体外中和病毒,同时也能保护小鼠和猪免受病毒感染[9]。因此,gC蛋白的B细胞表位也可用于亚单位疫苗的制备。gB、gC的中和抗体水平是评价免疫保护效率的重要指标之一。目前,基于PRV融合中和抗体的诊断试剂的研究较少,为此,本研究利用融合PCR技术将gB和gC片段融合(命名为gB-C),并利用基因工程方法原核表达gB-C重组融合蛋白,为猪群免疫水平检测及亚单位疫苗的制备奠定基础。
1 材料和方法
1.1 材料
1.1.1 细胞、载体及毒株 大肠杆菌(E.coli)DH5α感受态细胞购自天根生物公司,pMD19-T vector购自宝生物工程(大连)有限公司。PK-15细胞、PRV毒株、表达载体pET28a、BL21(DE3)感受态细胞均由农业部动物免疫学重点实验室保存。
1.1.2 主要试剂 DNA提取试剂盒、胶回收试剂盒、LATaqDNA聚合酶、限制性内切酶2×GC Buffer、BamHⅠ、HindⅢ、dNTP均购自宝生物工程(大连)有限公司。PRV阳性血清、His标签抗体(一抗)、羊抗猪IgG(二抗)均由农业部动物免疫学重点实验室保存。
1.1.3 引物设计 依据GenBank中PRV BJ/YT株gB和gC蛋白B细胞表位编码序列设计引物,由生工生物工程(上海)股份有限公司合成,引物序列为gB-C-P1:5′-CCGGGATCCCTTTGGCCTGCTCCACACCACGCTG-3′;gB-C-P2:5′-GCTCGCCCGC-GCTGCTGTTGCCGCCGGACGGCGGCGGGCAGACGT-AGAAGC-3′;gB-C-P3:5′-CCGGGATCCGGCGGCAACAGCAGCGCGGGCGAGC-3′;gB-C-P4:5′-CCCAAGCTTCTATTAGCGCGGGGGCTCGTCAA-AGTACTCG-3′。融合扩增片段长度为708 bp。
1.2 方法
1.2.1 PRV病毒基因组的提取 PK-15细胞形成单层后弃去生长液,接种200 μL PRV病毒液,在吸附2 h后加维持液,当70%细胞出现病变和脱落后,收集全部细胞培养物。在500 μL细胞培养物中,加入6 μL蛋白酶K、94 μL 10% (m/V)SDS,混匀,55 ℃加热30 min;接着将等体积酚-氯仿加入,颠倒混匀,12 000 r/min离心5 min;收集上清,再加入等体积氯仿,混匀,12 000 r/min离心5 min;将上清收集并加入2倍体积冷乙醇颠倒混匀,-20 ℃静置 10~20 min,12 000 r/min离心10 min;用70%的乙醇将沉淀洗涤1次,放到超净台使残留乙醇挥发干净;用25 μL含RNase (20 μg/mL)的灭菌去离子水溶解,尽快使用或置于-20 ℃条件下保存。
1.2.2 PRV病毒基因组的鉴定 根据参考文献[3]合成如下引物,用来鉴定伪狂犬病毒,p1:5′-CACGGAGGACGAGCTGGGGCT-3′;p2:5′-GTCCACGCCCCGCTTGAAGCT-3′。以病毒DNA作为模板,鉴定PCR反应体系为:模板DNA 1 μL,上、下游引物各1 μL,ExTaq预混酶10 μL,ddH2O 7 μL。扩增程序为:95 ℃ 5 min;94 ℃ 40 s,65 ℃ 30 s,72 ℃ 45 s, 31个循环;72 ℃ 10 min。取PCR产物进行1%琼脂糖凝胶电泳。
1.2.3gB-C融合表位编码序列制备 以提取的病毒DNA为模板,利用引物gB-C-P1、gB-C-P2进行gB糖蛋白B细胞表位编码序列的PCR扩增,利用引物gB-C-P3、gB-C-P4进行gC糖蛋白B细胞表位编码序列的PCR扩增。反应体系为20 μL:模板DNA 1 μL,LATaq酶0.2 μL,上、下游引物各0.5 μL,2×GC Buffer 10 μL,dNTP 1 μL,ddH2O 6.8 μL。混匀后置PCR扩增仪中,95 ℃预变性5 min;94 ℃ 30 s,69 ℃ 30 s,72 ℃ 45 s,30个循环;72 ℃延伸10 min。用上述方法分别获得gB和gC糖蛋白B细胞表位编码序列。取PCR产物进行琼脂糖凝胶电泳,用凝胶成像系统拍照记录结果并进行回收。然后将相同摩尔数的片段gB和gC混合为模板,上、下游引物分别为gB-C-P1、gB-C-P4,进行融合扩增获得糖蛋白B细胞表位融合编码序列。反应条件:95 ℃预变性5 min;94 ℃ 30 s,69 ℃ 30 s,72 ℃ 90 s,30个循环;72 ℃延伸10 min。取PCR产物进行琼脂糖凝胶电泳,用凝胶成像系统拍照记录结果并进行回收。
1.2.4 pMD19T-gB-C重组质粒的构建 将回收的DNA片段与PMD19-T载体16 ℃过夜连接,将10 μL连接产物加入到DH5α大肠杆菌感受态细胞中,轻轻混匀,冰浴30 min;42 ℃热休克90 s,迅速置冰上3 min,随后加入1 ml LB培养液,37 ℃ 220 r/min振荡培养1 h。3 000 r/min离心3 min,弃上清,留200 μL悬浮细胞沉淀,涂布含氨苄青霉素(100 μg/mL)的LB平板,37 ℃过夜培养至菌落出现,进行双酶切及质粒PCR鉴定,构建pMD19T-gB-C重组质粒。
1.2.5 表达载体的构建 将阳性质粒pMD19T-gB-C和表达载体pET28a分别用限制性内切酶BamHⅠ和HindⅢ双酶切,将酶切回收后的目的片段和表达载体用T4 DNA连接酶连接。将连接产物转化入DH5α感受态细胞中,挑取单克隆,构建质粒pET-28a-gB-C。对pET-28a-gB-C进行质粒PCR和双酶切鉴定。将鉴定为阳性的pET-28a-gB-C菌液送往生工生物工程(上海)股份有限公司进行序列测定。
1.2.6 诱导表达 将测序鉴定正确的重组阳性质粒转化BL21(DE3)感受态细胞,挑菌斑过夜培养,取重组菌液100 μL 接种于含100 μg/mL氨苄青霉素的100 mL LB培养液中,在温度37 ℃、230 r/min的摇床内培养至OD600为0.6~1.0,加入终浓度为1.0 mmol/L的IPTG,24 ℃诱导表达8 h。
1.2.7 表达产物的检测
1.2.7.1 SDS-PAGE电泳 配置12%的分离胶。取诱导表达菌液1 mL于1.5 mL 离心管内,12 000 r/min离心2 min。弃上清,用200 μL PBS重悬,加入50 μL 5×Buffer(用前加热)。放入浮漂中,煮沸5 min后,12 000 r/min离心2 min。将未诱导的重组菌作为对照。取20 μL上清上样至电泳槽内,电泳后放入考马斯亮蓝染色液中染色3 h,然后脱色观察。
1.2.7.2 Western blot检测 取NC膜,剪成比蛋白胶稍大的长方形,之后按照滤纸→NC膜→SDS胶→滤纸的顺序放入电转仪(膜与膜之间无气泡),15 V电转1 h。电转后的NC膜用5%的脱脂奶粉4 ℃封闭过夜,依次加入一抗、二抗进行处理,最后用AEC显色方法进行显色。
2 结果与分析
2.1 PCR扩增结果
提取伪狂犬病毒DNA,以此为模板,应用特异性引物PCR扩增,将PCR产物进行1%琼脂糖凝胶电泳,紫外灯光下观察,可见PCR扩增出的片段大小约为217 bp,与预期结果一致(图1)。
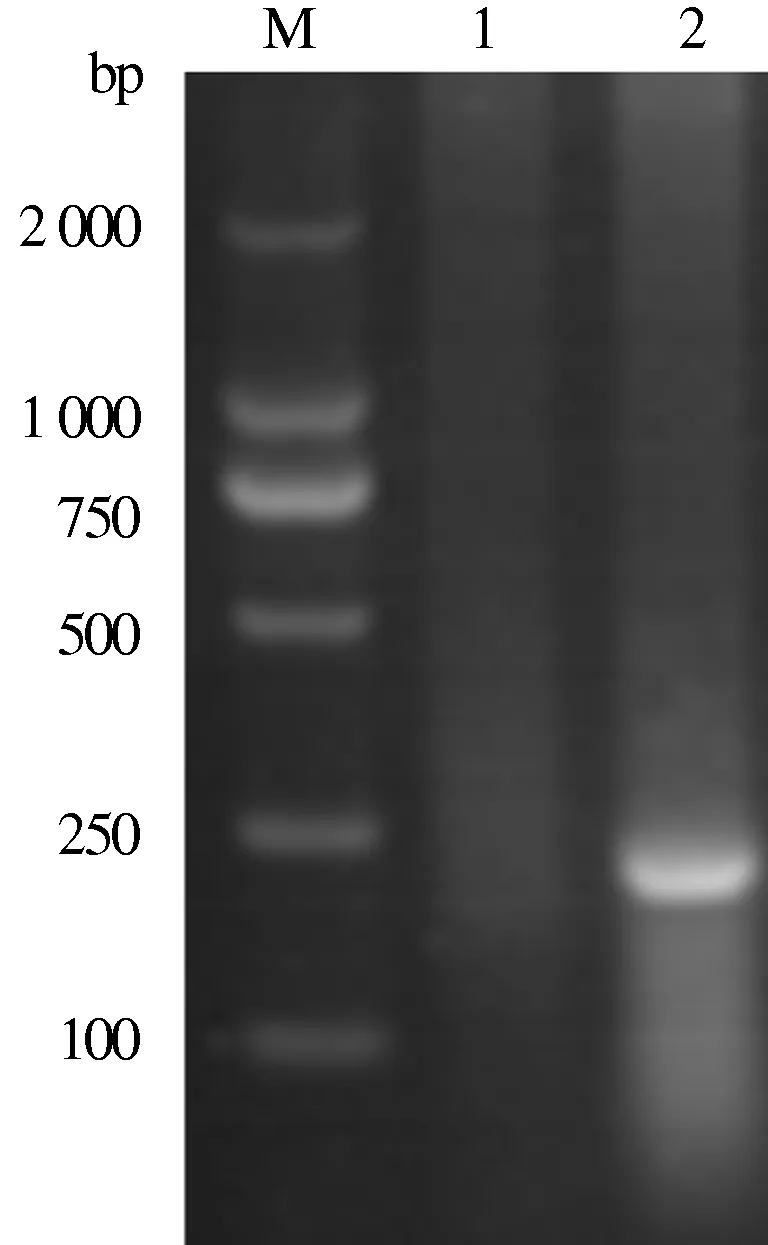
M:DNA Marker; 1:正常细胞DNA 模板扩增;2:病毒培养物DNA 模板扩增图1 PRV DNA模板PCR鉴定
以提取的病毒DNA为模板,利用引物gB-C-P1、gB-C-P2进行扩增,获得了与设计大小(346 bp)一致的gB蛋白B细胞表位编码序列的特异条带(图2)。利用引物gB-C-P3、gB-C-P4进行扩增获得387 bp的gC蛋白B细胞表位编码序列的特异性条带(图3)。以扩增出的gB、gC表位编码序列片段为模板,利用引物gB-C-P1、gB-C-P4对其进行融合扩增,获得了与预计大小一致的708 bp的gB-C蛋白B细胞表位融合编码序列的特异条带(图4)。
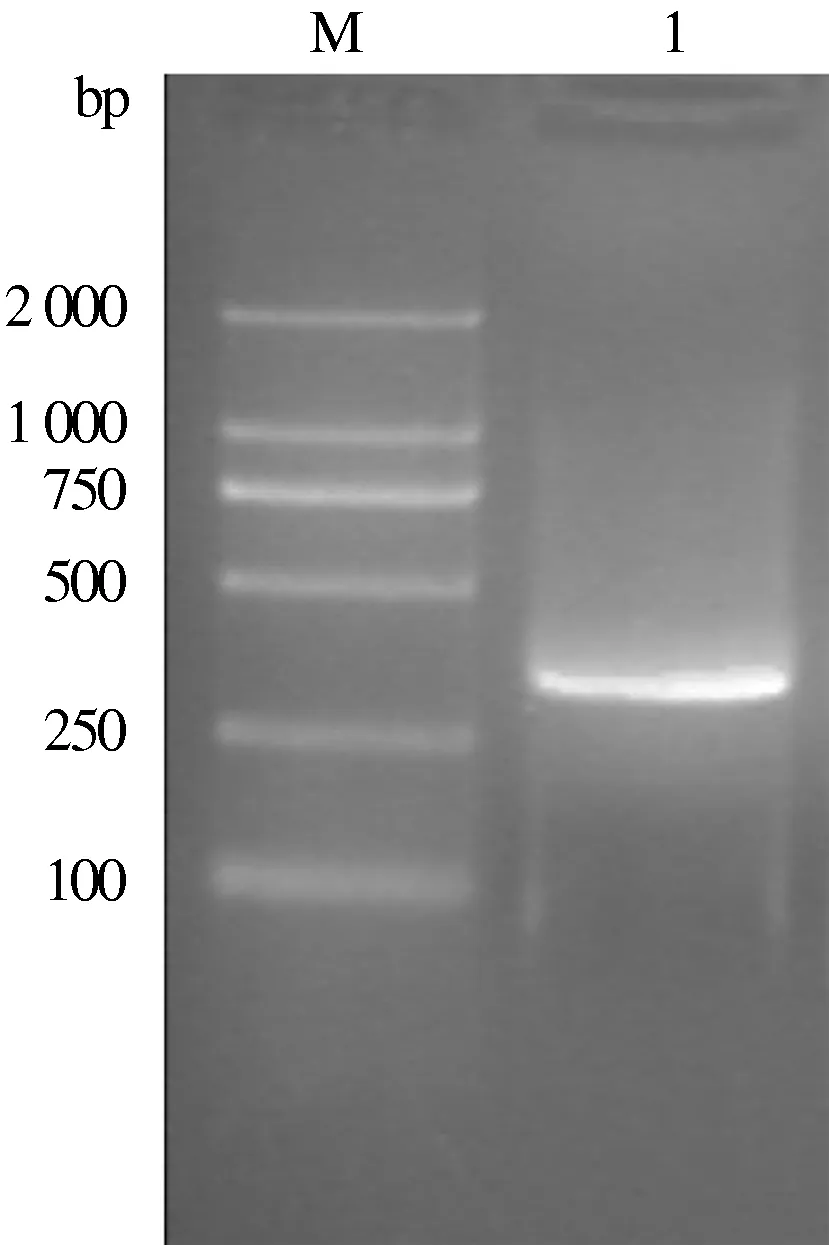
M:DNA Marker;1:gB蛋白B细胞表位编码序列扩增片段图2 gB蛋白B细胞表位编码序列扩增结果
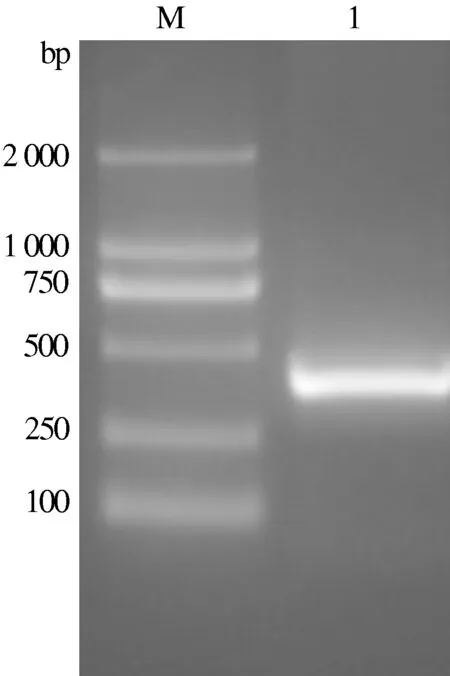
M:DNA Marker;1:gC蛋白B细胞表位编码序列扩增片段图3 gC蛋白B细胞表位编码序列扩增结果
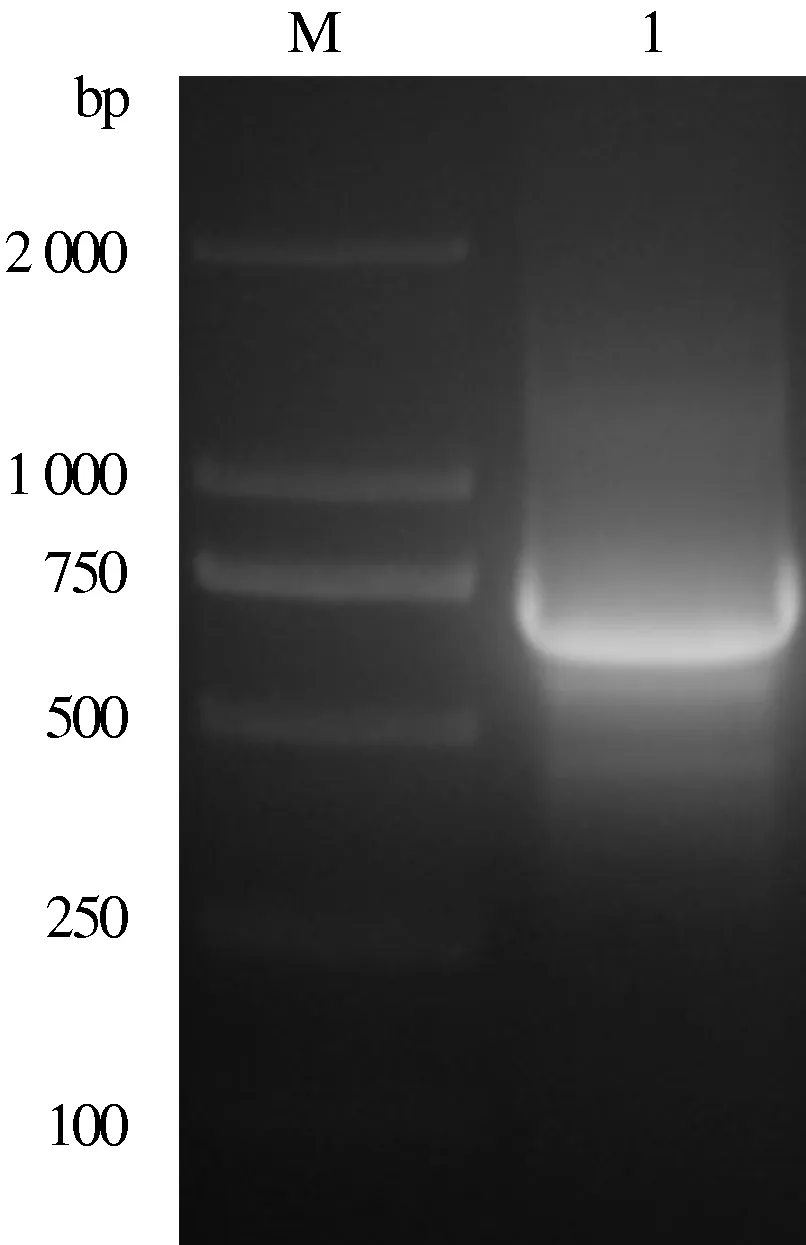
M:DNA Marker; 1:gB-C融合蛋白B细胞表位编码序列片段图4 gB-C融合蛋白B细胞表位编码序列扩增结果
2.2 pET-28a-gB-C的鉴定结果
2.2.1 酶切鉴定 对重组质粒pET-28a-gB-C用限制性内切酶BamHⅠ、HindⅢ酶切后,经琼脂糖电泳可见目的条带708 bp 和载体条带5 900 bp,与预计的一致(图5)。
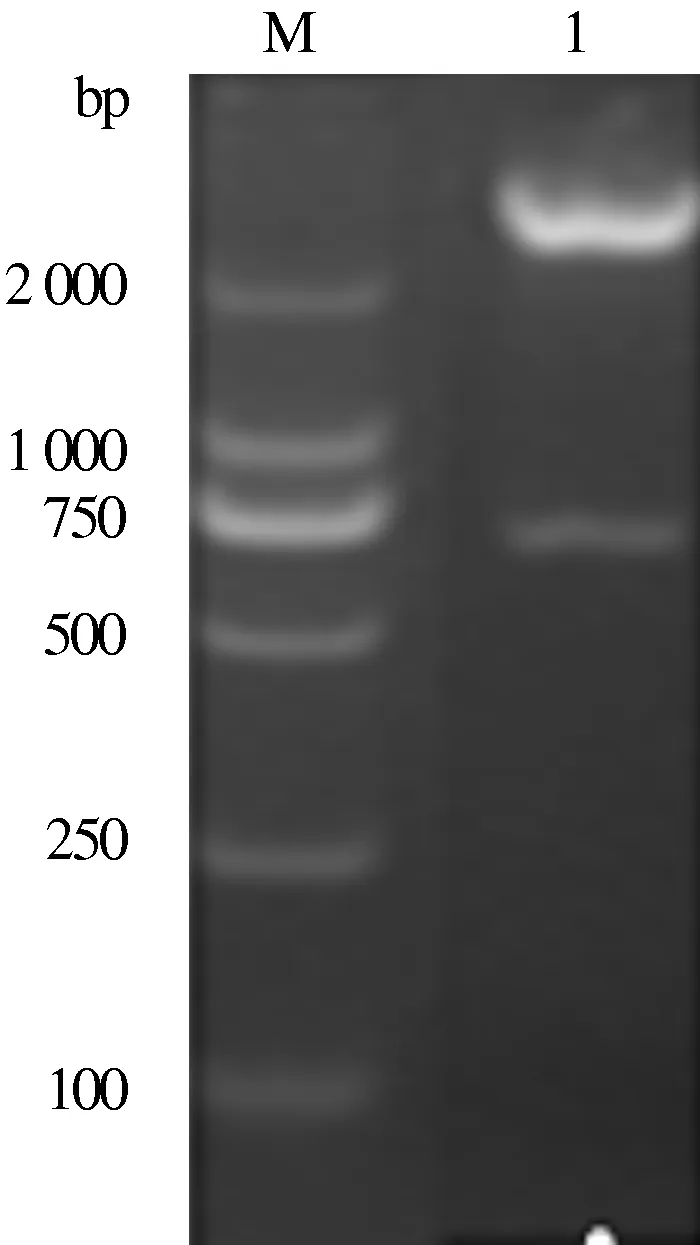
M:DNA Marker; 1:pET-28a-gB-C质粒图5 pET-28a-gB-C质粒双酶切鉴定结果
2.2.2 PCR鉴定 以初步鉴定为阳性的重组质粒为模板,用引物gB-C-P1、gB-C-P4按上述扩增条件进行PCR扩增,扩增出了大小约700 bp的条带,与预期结果相符(图6)。

M:DNA Marker;1:gB-C编码序列片段图6 pET-28a-gB-C质粒PCR鉴定结果
2.2.3 序列测定 将阳性重组质粒进行序列分析,插入的基因阅读框架无误,测序结果与预期结果一致(测序图表略)。
2.3 表达产物的鉴定
2.3.1 SDS-PAGE鉴定 重组阳性菌用IPTG在24 ℃诱导表达后,经SDS-PAGE电泳检测,结果显示,诱导的重组菌比未诱导重组菌多出1条分子质量大小约38 ku的目的蛋白条带,与融合蛋白预期大小一致(图7)。由此证明,融合的目的蛋白在原核系统中成功表达。
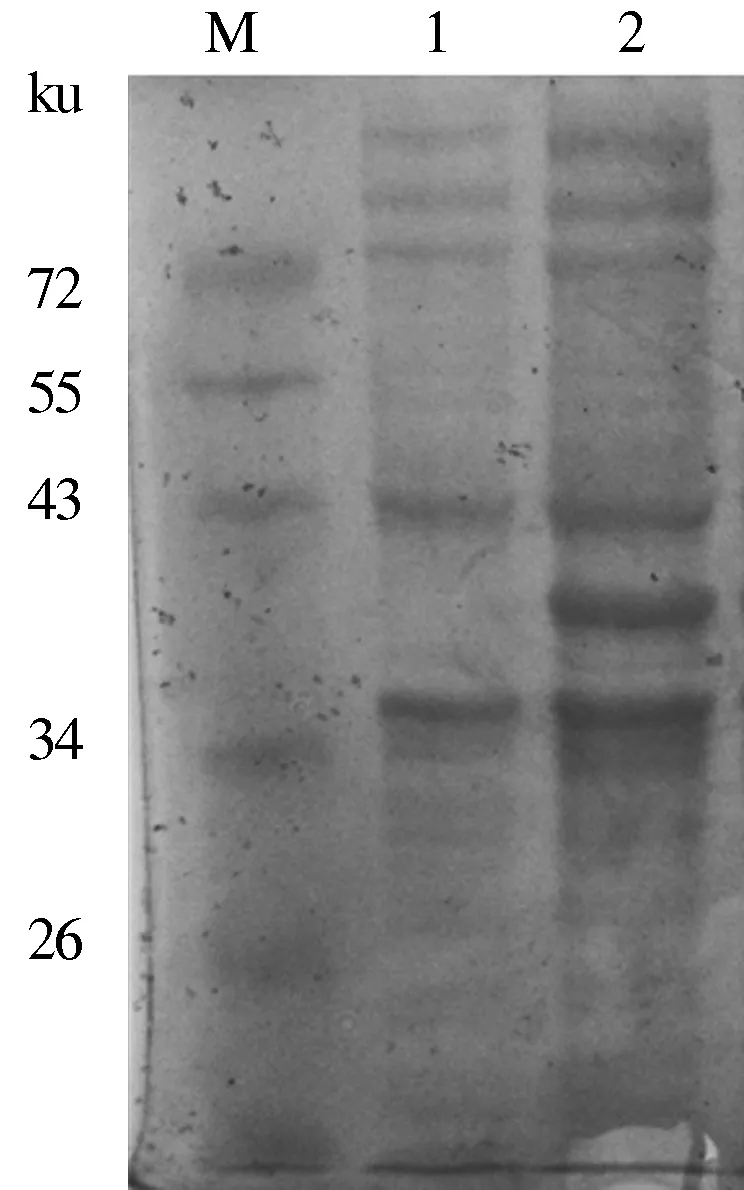
M:预染Marker;1:pET-28a-gB-C未诱导组; 2:pET-28a-gB-C诱导组图7 gB-C融合蛋白的SDS-PAGE分析结果
2.3.2 Western blot分析 表达产物经SDS-PAGE电泳后,将蛋白质条带转移到NC膜上进行免疫印迹分析。结果显示,在约38 ku大小的位置出现特异性反应条带(图8)。结果证实,表达的融合表位能够被阳性血清识别。
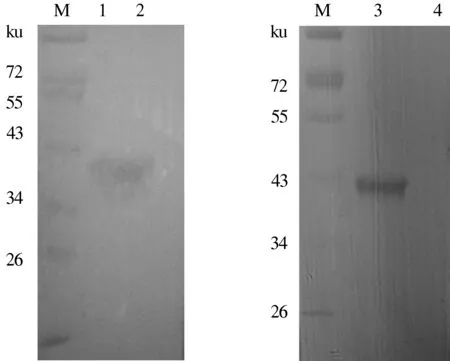
M:预染Marker; 1:阳性血清诱导结合组; 2:阳性血清未诱导结合组; 3:His标签蛋白诱导结合组; 4:His标签蛋白未诱导结合组图8 gB-C融合蛋白的Western blot鉴定结果
3 结论与讨论
猪伪狂犬病对我国养猪业的健康发展造成了极大威胁,猪群病毒净化、提高免疫密度和免疫强度、定期检测猪群的免疫水平,是目前控制此病的有效方法,其中免疫监测尤为重要[10-15]。本试验将PRVgB、gC表位基因应用融合PCR技术连接,采用李敏等[16]的一步融合PCR方法不能得到预期的融合片段,而应用Davidson等[17]两步 PCR方法,获得融合基因gB-C。将融合的gB-C基因片段克隆到原核表达载体pET28a,采用大肠杆菌BL21(DE3)表达系统, IPTG浓度由0.2 mmol/L升高至1.0 mmol/L,诱导温度由37 ℃降低至24 ℃,诱导时间由2 h提高至8 h,得到的目的蛋白能够识别PRV阳性血清,表明该蛋白具有较好的免疫学活性。
本研究将gB、gC糖蛋白B细胞表位进行融合克隆表达,获得了高纯度的检测抗原,提高了抗体检测的敏感性、特异性和准确性,为制备用于PRV抗体检测的试纸条和ELISA检测试剂盒奠定了基础。
[1] 杨庆芳,宁关宝.猪伪狂犬病的研究进展[J].畜牧兽医科技信息,2010,7(7):15-18.
[2] 母安雄,谷根林,蒋建一,等.规模猪场主要疫病抗体水平监测及免疫效果分析[J].畜牧与兽医,2002,34(3):29-31.
[3] 周复春,陈焕春,李学伍,等.PCR技术检测猪伪狂犬病毒及其潜伏感染部位的研究[J].动物医学进展,1997,18(2):22-25.
[4] Zaripov M M,Morenkov O S,Fodor N,etal.Distribution
of B-cell epitopes on the pseudorabies virus glycoprotein B[J].Journal of General Virology,1999,80(Pt3):537-541.
[5] Zaripov M M,Morenkov O S,Siklodi B,etal.Glycoprotein B of Aujeszky’s disease virus:Topographical epitope mapping and epitope-specific antibody response[J].Research in Virology,1998,149:29-41.
[6] Rue C A,Ryan P.Pseudorabies virus glycoprotein C attachment-proficient revertants isolated through a simple,targeted mutagenesis scheme[J].Journal of Virological Methods,2008,151(1):101-106.
[7] Rue C A,Ryan P.A role for glycoprotein C in pseudorabies virus entry that is independent of virus attachment to heparan sulfate and which involves the actin cytoskeleton[J].Journal of Virology,2003,307(1):12-21.
[8] Ober B T,Teufel B,Wiesmüller K H,etal.The porcine humoral immune response against pseudorabies virus specifically targets attachment sites on glycoprotein gC[J].Journal of Virology,2000,74(4):1752-1760.
[9] Riviere M,Tartaglia J,Perkus M E,etal.Protection of mice and swine from pseudorabies virus conferred by vaccinia virus-based recombinants[J].Journal of Virology,1992,66(6):3424-3434.
[10] 樊振华,姚敬明,孟帆,等.山西部分种猪场猪伪狂犬病分子流行病学调研[J].山西农业科学,2012,40(9):989-992.
[11] 杨庆芳,宁官保,李俊达.猪伪狂犬病病毒的分离鉴定[J].山西农业科学,2011,39(8):886-889.
[12] 余波,周思旋,谭诗文,等.猪伪狂犬病毒野毒株SYBR Green Ⅰ实时荧光定量PCR诊断试剂盒的研制[J].河南农业科学,2014,43(6):128-131,144.
[13] 高晓云,顾阳,潘鑫龙,等.猪伪狂犬病病毒河南分离株gE全基因的克隆与序列分析[J].华北农学报,2015,3(1):137-141.
[14] 赵朴,郑玉姝,赵坤,等.猪呼吸道疾病综合征常见病毒多重PCR方法的建立及应用[J].华北农学报,2014,29(3):64-67.
[15] 顾阳,高晓云,程琨,等.鉴别猪伪狂犬病病毒强毒与疫苗毒双重PCR检测方法的建立[J].华北农学报,2014,29(2):94-97.
[16] 李敏,杨谦.一种高效构建同源重组 DNA片段的方法融合——PCR[J].中国生物工程杂志,2007,27(8):53-58.
[17] Davidson R C,Blankenship J R,Kraus P R,etal.A PCR obased strategy to generate integrative targeting alleles with large regions of homology[J].Microbiology,2002,148(Pt8):2607-2615.
Fusion Expression and Activity Detection of Porcine Pseudorabies Virus gB and gC Glycoprotein B-cell Epitope Coding Sequences
ZHANG Chong1,2,LIU Fang1,ZHAO Dong2,DENG Ruiguang2,ZHANG Gaiping2,3,LI Xuewu2*
(1.College of Animal Science and Technology,Henan Science and Technology University,Luoyang 471003,China; 2.Henan Provincial Key Laboratory of Animal Immunology,Henan Academy of Agricultural Sciences,Zhengzhou 450002,China; 3.College of Veterinary Medicine and Animal Science,Henan Agricultural University,Zhengzhou 450002,China)
According to PRV B cell epitope sequences of the gB and gC protein,the fusion primers were designed respectively.Using the fusion PCR technology,the gB-Cwas fused by the B cell epitope sequences of the gB and gC protein.Then the gB-Cwas inserted into the pET28a.The recombinant plasmid was transformed intoE.coli(BL21).TheE.coli(BL21)expressed the recombinant protein which was induced with 1 mmol/L IPTG.The result of SDS-PAGE showed that the gB-C was expressed successfully.The molecular weight of the recombinant protein was 38 ku.The result of Western blot showed that the recombinant gB-C protein could be recognized by His-tag mouse Mcab and PRV porcine positive serum.In this research,the recombinant gB-C protein was expressed successfully and the recombinant protein had a good immunological activity.
PRV; gB protein; gC protein; B-cell epitope; fusion expression
2015-07-10
国家自然科学基金项目(31172348)
张 冲(1989-),男,河南新乡人,在读硕士研究生,研究方向:分子病原学和免疫学。E-mail:421986388@qq.com
*通讯作者:李学伍(1964-),男,河南通许人,研究员,博士,主要从事动物病原学和免疫学研究。 E-mail:lixuewu2002@126.com
S855.3
A
1004-3268(2016)01-0119-05
猜你喜欢
广东药科大学学报(2022年3期)2023-01-04
生物学通报(2022年1期)2022-11-22
江西农业学报(2021年4期)2021-04-20
温州医科大学学报(2019年4期)2019-04-28
罕少疾病杂志(2017年2期)2017-02-23
中国免疫学杂志(2017年1期)2017-01-17
广西林业科学(2016年3期)2016-03-16
西南医科大学学报(2015年1期)2015-08-22
畜牧兽医学报(2015年3期)2015-07-05
医学研究杂志(2015年6期)2015-07-01
