
HIV-1逆转录酶抑制剂的3D-QSAR研究及分子设计
2016-04-13 09:13仝建波吴英纪
分析测试学报 2016年11期
仝建波,占 培,吴英纪
(陕西科技大学 化学与化工学院,陕西 西安 710021)
HIV-1逆转录酶抑制剂的3D-QSAR研究及分子设计
仝建波*,占 培,吴英纪
(陕西科技大学 化学与化工学院,陕西 西安 710021)
采用Topomer CoMFA方法对24个二芳基苯胺衍生物进行三维定量构效关系研究,建立了3D-QSAR模型,所得优化模型的非交叉相关系数、交互验证系数以及外部验证的复相关系数分别为0.928,0.654和0.940,结果表明该模型具有良好的稳定性和预测能力。采用分子对接技术对药物与受体的作用机制进行了研究,结果显示,药物与HIV-1逆转录酶的LYS172,GLU138,LYS101等位点作用明显。运用这些信息进行分子设计,在理论上获得了一些具有较高活性的新的二芳基苯胺类抗艾滋病药物,该QSAR的研究结果可为新药合成提供理论参考。
三维定量构效关系;易位体比较分子场;分子对接;分子设计;二芳基苯胺衍生物
获得性免疫缺陷综合症(Acquired immune deficiency syndrome,AIDS),即艾滋病病毒,是造成人类免疫缺陷病毒(Human immunodeficiency virus HIV)的一种致命疾病,HIV(分为HIV-1和HIV-2型)属逆转录病毒的一种,通过破坏人体的T淋巴细胞(CD4)、感染单核细胞和巨噬细胞,然后经过吸附、融合过程进人宿主细胞,进而阻断细胞免疫和体液免疫过程,可导致机体免疫功能部分或全部丧失,继而发生机会性感染肿瘤等[1]。HIV 病毒密码是由逆转录酶、蛋白酶和整合酶3种关键酶来表达[2],近几年来抗HIV/AIDS药物的研制主要是针对抑制上述关键酶进行的,研制这些酶的抑制剂成为治疗艾滋病的一种有效手段[3]。
二芳基苯胺衍生物是一种典型的抗艾滋病药物,对于病毒逆转录酶表现出较高的抑制活性,如果通过优化结构使得活性提高,则二芳基苯胺衍生物将会成为一种高效的抗艾滋病药物。定量构效关系(Quantitative structure activity relationship,QSAR)是基于配体的计算机辅助药物设计的主要方法之一,具有代表性的方法有CoMFA[4]与CoMSIA[5]。本实验采用第二代CoMFA 方法——Topomer CoMFA[6-7]对24种二芳基苯胺衍生物进行了3D-QSAR的研究,并进一步采用分子对接技术模拟二芳基苯胺衍生物与HIV-1逆转录酶之间的结合构象,按照几何互补、能量互补、化学环境互补的原则来评价配体与受体的相互作用,并找到两个分子之间最佳的结合模式,从而在设计新的拟制剂时以结合模式最佳的化合物进行合成[8]。运用这些研究结果,进行分子设计,提出一些可能具有较高抗艾滋病活性的新的二芳基苯胺衍生物,为实验工作者合成该系列新药物提供理论参考。
1 实验部分
1.1 易位体比较分子场
Topomer CoMFA是CoMFA与Topomer的联合技术,能够在短时间内准确、快速构建3D-QSAR模型。采用Topomer技术将整个配体分子切割成两个或两个以上的小片段,所切割形成的小片段会自动生成三维构象的碎片,碎片根据一定的经验规则进行调整,生成Topomer模型[9]。计算每个分子片段周围的立体场和静电场参数,作为自变量,以EC50的负对数(pEC50)[10]为建模响应值,其中EC50值为 HIV-1病毒感染细胞半数抑制浓度,用偏最小二乘回归分析方法[11]寻找这些三维结构的特征信息与化合物活性的联系,实现模型拟合。
1.2 数据来源与分子结构构建
从文献[12]中搜集了具有确定活性的24个二芳基苯胺衍生物(见表1)作为研究对象,其化学结构见图1,活性标度为pEC50(-logEC50)。从文献中列举的一系列化合物中随机选择18个化合物作为训练集(Training set)用于建立模型,剩余6个分子作为测试集(Test set)用来验证模型的外部预测能力。利用SYBYL 2.0-X软件包中的Sketch Molecule模块绘制出化合物的结构,以分子动力学程序 Minimize 对所有化合物进行能量优化,得到其最低能量构象。优化过程中采用Tripos力场[13]、powell 能量梯度和加载Gasteiger-Huckel电荷,迭代系数设置为2 000 次,能量收敛限定设为0.5 kcal/mol,并将RMS(均方根)设为0.005,其余参数采用SYBYL2.0-X的默认值[14]。
1.3 分子对接
对接软件是由AutoDock 4.2,HIV-1逆转录酶的三维结构从蛋白数据库下载,PDB ID为1S6Q,在进行对接之前除去原小分子配体及所有的水分子,给蛋白质加氢并计算Gasteiger电荷,使用SYBYL2.0-X构建配体结构式并进行优化。分子对接的格点为40×40×40,格点间距为0.375 nm,对接活性中心为X:147.806,Y:-26.833,Z:73.028。配体构象的搜索过程使用拉马克遗传算法(Lamarckian GA),配体与受体间的能量匹配通过半经验自由能的计算方法进行评价[15]。每个抑制剂小分子运算循环次数设置为50,能量评估的最大次数为250万次,其他参数为默认值。对接后会产生10个对接构象,根据各构象的结合模式和结合自由能选择合适的构象进行研究,计算采用如下的函数形式。抑制剂和氨基酸残基的相互作用分为5项,即范德华相互作用、静电相互作用、可旋转键能、氢键相互作用和去溶剂化能[16]。
方程式中ΔGvdw,ΔGH-bond,ΔGele,ΔGtor和ΔGsol均为半经验参数,Ntor指配体在对接后被约束的可旋转键的数目。
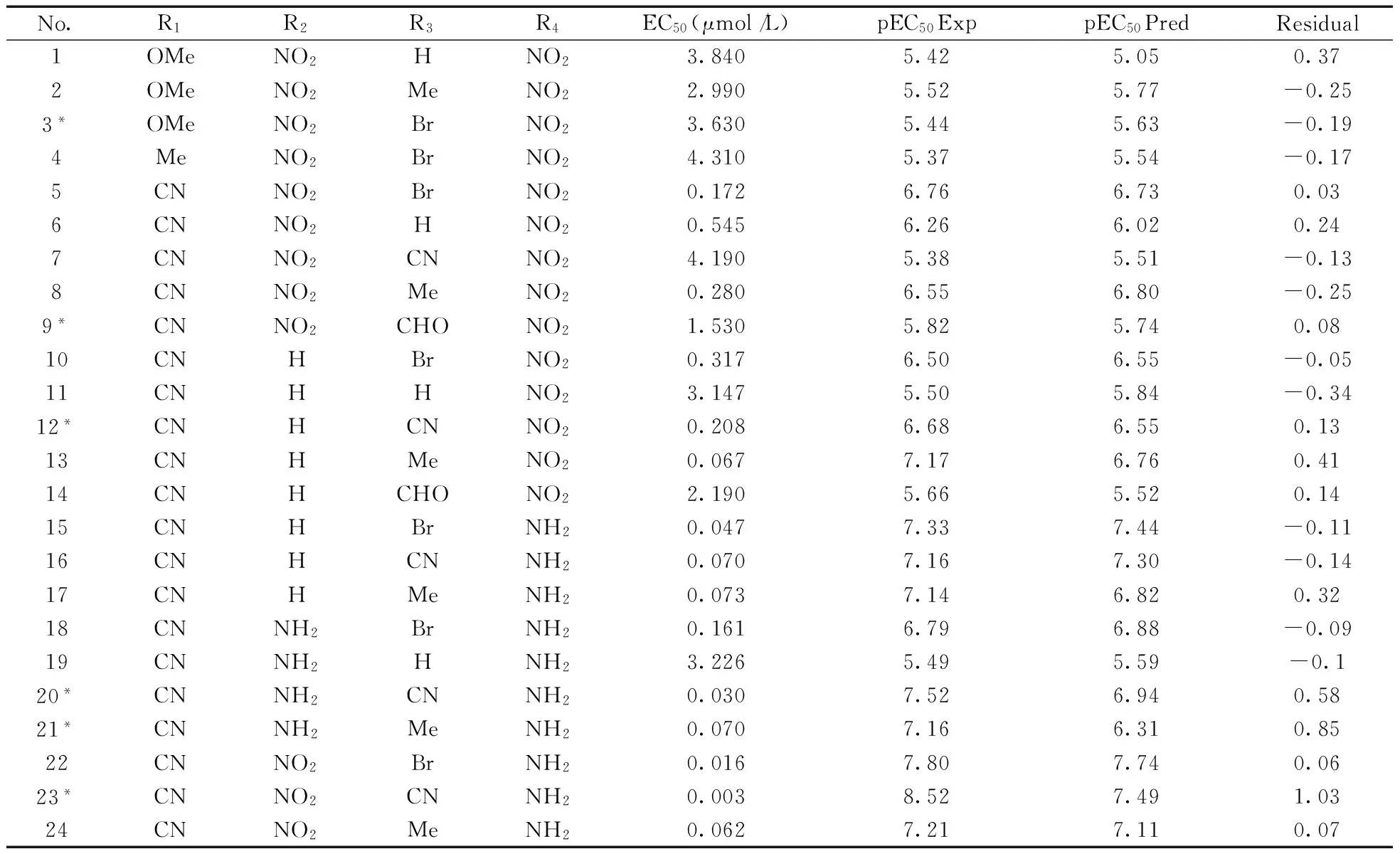
表1 化合物的结构及活性数据Table 1 The structure and biological activity of the compound
*:test sets(测试集)
2 结果与讨论
2.1 Topomer CoMFA模型的统计结果及预测能力

2.2 3D-QSAR分析
图3为以22号分子为模板的Topomer CoMFA模型立体场和静电场的三维等势图,其中图3A和C分别为Ra,Rb的立体场三维等势图,图3B和D分别为Ra,Rb的静电场三维等势图,图3A和C中绿色区域表示引入大体积的基团有利于分子抑制活性,而黄色区域代表引进小体积的基团有利于化合物抑制活性增加;图3B和D中蓝色区域表示引入带正电荷的基团有利于分子抑制活性,而红色区域代表引进带负电荷的基团有利于化合物抑制活性增加。由图3A立体场等势图中可观察到在R3取代基上有绿色区域,图3C立体场等势图中可观察到在R1取代位点上也有绿色区域,表明在这2个区域中,将大分子基团引于此可以增加分子的活性。如将21,8号分子的R3基团以较大的基团—CH3取代了19,6号分子R3基团上较小的取代基—H,其活性有明显提高,3,10,18,5号分子的R3基团以较大的基团-Br取代了1,11,19,6号分子R3基团上较小的取代基—H,其活性均明显提高。在R1取代基上3号分子以较大的基团—OCH3取代了4号分子R3基团上较小的取代基—CH3,其活性明显提高。
由图3D静电场等势图可知,在临近切割处环绕着一块蓝色等势域,表明若此区域有正电性基团作用,可增加化合物活性。在R1取代基上可以看到R1被一大型的红色轮廓包裹,表明在此区域若引入带负电的取代基可促使化合物活性增加,如5号分子的R1取代基由—CN取代了4号分子的—CH3,生物活性由5.37提高至6.76,3号分子R1取代基上由—OCH3取代4号分子的—CH3,其活性也由5.37提高至5.44。图3B中,R2和R4取代基上聚有蓝色等势域,表明在此区域若引入带正电的取代基可促使化合物活性增加,如12和17号分子的R2基团以—H取代了7和8号分子的取代基—NO2,其活性相对提高。24,23,22,15,17号分子的R4基团以—NH2取代了8,7,5,10,13号分子的取代基—NO2,其活性均相对提高。
根据以上分析,若要增加化合物活性,可在绿色区域引入大基团或在黄色区域引进小分子基团,而在红色区域引入负电荷基团或在蓝色区域引入正电荷基团。
2.3 分子对接研究
2.3.1 分子对接方法预测能力的评估 在生成模型之前,验证分子对接方法的可靠性是非常重要的。通过AutoDock将配体还原对接到1S6Q蛋白酶结合口袋,验证对接方法能否再现结合底物在晶体结构中的构象[17-18]。对接后的分子构象与晶体结构中配体分子构象的均方根偏差(RMSD)作为评价标准[19],一般RMSD在0.029 2~0.215 4 nm之间即认为对接还原成功,认为对接构象在结合位点内与原晶体构象是相似的。本实验选择3个结合能量低的对接构象进行验证分析,3个构象的RMSD均小于0.215 4 nm(分别为0.210 9,0.207 4,0.194 5 nm),对接结果如图4所示,表明上述对接方法能成功在晶体结构中还原再现结合底物的构象。2.3.2 对接结果与分析 本文选取活性最高的23号化合物与1S6Q蛋白酶进行分子对接研究(见图5),以结合自由能最低的构象进行分析。在药物与受体的作用模式中,氢键是药物分子和受体生物大分子之间较为普遍的一种键合方式,可以增加药物的活性。
图5为化合物与1S6Q蛋白酶的对接作用模式图,可以看出化合物与1S6Q蛋白酶受体活性部位氨基酸残基形成了氢键(图5A),并可观察到氢键的给体和受体(图5B)。化合物与1S6Q蛋白酶受体活性部位氨基酸残基LYS172上H原子与苯环上连接的—CN上的N原子形成氢键,距离为2.175 1 Å,其次氨基酸残基LYS101尾端两个O原子和GLU138尾端两个H原子也形成氢键,距离分别为1.596 9 Å和2.094 9 Å。氢键起到增加小分子与蛋白酶结合强度以及活性中心小分子的定向作用,这些力将小分子稳定在酶活性中心由ILE180,THR139,VAL179,PHE229,VAL106,LEU100,LYS172,GLY190,LYS101,TYR188,LEU234,LYS103所构成的口袋中。由于氢键的存在,所以5号化合物的活性高于4号和3号,8号化合物的活性高于2号,6号化合物的活性高于1号,这与Topomer CoMFA模型分析所得的结论一致。
2.4 基于研究结果的分子设计
研究结果表明,取代基R1和R3被大体积基团取代,在R1的部位上引入带负电的取代基,在R2和R4上引入正电荷取代基,均有利于提高化合物的活性。以活性最高的化合物23为母体进行结构修饰,由于化合物23中R1和R3部位上取代基的大小适中,所以在静电效应R2取代基上进行结构修饰。根据极性交替规律和极性叠加原理[20-22],设计了3个使R2第1个原子具有大的净正电荷的基团及相应的化合物。设计基团其原子间的极性示意图如图6所示。

CompoundR1R2R3R4pEC50aOMeCOFHNO28.11bOMeCONO2MeNO28.55cOMeCFNO2BrNO28.60
由图6可见,与基团相连接的第1个C原子的箭头均为离开C1原子(其中实箭头表示直接的键极性方向,虚箭头表示由极性交替规律得到的诱导的键极性方向),由此可见C1的静正电荷必定很大,这与计算得到的结果相符。表2列出了由本模型计算得到的相应化合物的活性,其pIC50值为8.11~8.60,由此可见所设计的化合物具有高活性。
将设计好的3个化合物用于分子对接(如图7),从图7A中可以看出,化合物a与1S6Q蛋白酶受体活性部位氨基酸残基形成了多个氢键。氢键距离分别为1.762 7 Å(LYS101/—O…H—N—),2.021 5 Å(LYS101/—O…H—NH—),1.894 3 Å(LYS101/—N…H—NH—)。化合物b与1S6Q蛋
白酶受体活性部位氨基酸残基LYS101分别形成了3个氢键(图7B),氢键作用部位与化合物a相同,氢键的距离分别为1.711 4 Å,2.117 8 Å,2.371 9 Å。图7C为化合物c的分子对接图,可以观察到1个氢键,其距离为2.175 2 Å(LYS172/—H…—CN)。图7表明所设计的3种化合物不仅有较高的活性,而且与蛋白酶的结合能力较强,进一步说明设计的3种化合物合理可信,能够为新药的合成提供理论指导。
3 结 论
本文利用分子对接和Topomer CoMFA方法对24个二芳基苯胺衍生物与1S6Q蛋白酶的作用模式进行了研究,建立了3D-QSAR模型,模型等势面图提供了立体场和静电场的可视化图像,直观地揭示了这一系列化合物中不同的取代基结构对其生物活性的影响,通过对接还原配体结晶构象验证了分子对接方法的准确性。结果表明,对接方法可以预测出正确的结合模式。采用分子对接从分子水平上阐述了二芳基苯胺衍生物与1S6Q蛋白酶的结合机制,并理论上提出了一些具有较高活性的二芳基苯胺衍生物,从而为实验工作者合成新药提供了理论参考。
[1] Fauci A S.Science,1988,239(4840):617-622.
[2] Tong J B,Wang P,Li Y F,Liu S L,Meng Y L.J.At.Mol.Phys.(仝建波,王平,李云飞,刘淑玲,孟元亮.原子与分子物理学报),2011,28(1):41-46.
[3] Young S D,Britcher S F,Tranl L O,Payne L S,Lumma W C,Lyle T A,Huff J R,Anderson P S,Olsen D B,Carroll S S,Pettibone D J,O'brien J A,Ball R G,Balani S K,Lin J H,Chen I W U,Schleif W A,Sardana V V,Long W J,Byrnes V W,Emini E A.Antimicrob.AgentsChemother.,1995,39(12):2602-2605.
[4] Cramer R D,Bunce J D,Patterson D E.J.Am.Chem.Soc.,1988,110(18):5959-5967.
[5] Klebe G.Perspect.DrugDiscov.,1998,3(12):87-104.
[6] Cramer R D.J.Med.Chem.,2003,46(3):374-388.
[7] Richard D C,Phillip C,Gunther S,William C C,Brian C,Brian B M,Farhad S.J.Chem.Inf.Model,2008,48(11):2180-2195.
[8] Aarti G,Rupinder T,Gajendra P S R.BMCBioinf.,2010,11(1):125-137.
[9] Wold S,Sjostrom M,Eriksson L.Chemom.Intell.Lab.Syst.,2011,58:109-130.
[10] Zhang Q Q,Yao Q Z,Zhang S P,Bi L M,Zhou Z G,Zhang J.J.ActaPhys-Chim.Sin.(张青青,姚其正,张生平,毕乐明,周之光,张骥.物理化学学报),2014,30(2):371-381.
[11] Shui P X,Sun Q,Zhuang Y C,He B.J.Chin.Tradit.Med.(税丕先,孙琴,庄元春,何兵.中成药),2007,29(12):1870-1872.
[12] Kamlendra S B,Mukesh C S,Shailesh V J.J.TaibahUniv.Sci.,2015,9(4):521-530.
[13] Clark M,Cramer R D,Opdenbosch V N.J.Comput.Chem.,1989,10(8):982-1012.
[14] Liu Y L,Li Y T,Shi Z B,Zhong K,Shao Y Q,Zeng Y F,Huang D D,Wang G X,Liang G Z.Sci.Sin.:Chim.(刘永澜,李月婷,史博智,钟刊,邵奕强,曾亚飞,黄丹丹,王贵学,梁桂兆.中国科学:化学),2013,43(2):198-208.
[15] Zhang J L,Hou R Z,Li Z,Zheng Q C,Zhang H X.ActaChim.Sin.(张继龙,侯瑞哲,李卓,郑清川,张红星.化学学报),2010,68(3):222-226.
[16] Huey R,Morris G M,Olson A J,Goodsell D S.J.Comput.Chem.,2007,28(6):1145-1152.
[17] Shen M Y,Zhou S Y,Li Y Y,Pan P C, Zhang L L,Hou T J.J.Mol.Biosyst.,2013,9:361-374.
[18] Shen M Y,Yu H D,Li Y Y,Li P X,Pan P C,Zhou S Y,Zhang L L,Li S,Lee S M Y,Hou T J.Mol.Biosyst.,2013,9(6):1511-1521.
[19] Liu H C,Lu S,Ran T,Zhang Y M,Xu J X,Xiong X,Xu A Y,Lu T,Chen Y D.ActaPhys-Chim.Sin.(刘海春,卢帅,冉挺,张艳敏,徐金星,熊潇,徐安阳,陆涛,陈亚东.物理化学学报),2015,31(11):2191-2206.
[20] Pople J A,Gordon M.J.Am.Chem.Soc.,1967,89(17):4253-4265.
[21] Zheng K C,He F,Xu Z T,Yun F C.ActaPhys-Chim.Sin.(郑康成,何峰,许植涛,云逢存.物理化学学报),1999,15(8):698-703.
[22] Zheng K C,Wang J P,Peng W L,Liu X W,Yun F C.J.Phy.Chem.A,2011,105(48):10899-10905.
3D-QSAR and Molecular Docking Studies on Diarylaniline Derivatives of HIV-1 Reverse Transcriptase
TONG Jian-bo*,ZHAN Pei,WU Ying-ji
(College of Chemistry and Chemical Engineering,Shaanxi University of Science & Technology,Xi'an 710021,China)
Topomer CoMFA was used to build a three-dimensional quantitative structure-activity relationship(3D-QSAR) model for 24 diarylaniline derivatives in this paper.The coefficient of cross validation,the coefficient of non-cross validation and external validation were 0.654,0.928 and 0.940,respectively.The result showed that this model had a good stability and a predictive ability.By using molecular docking,the action mechanism of drug and acceptor was studied,and the results showed that the drug functions obviously with LYS172,GLU138,LYS101 sites of HIV-1 reverse transcriptase.Based on the above informations,three new molecules of diarylaniline derivatives with high activity were theoretically designed,and the QSAR results could offer an atheoretical reference for the pharmaceutical synthesis.
3D-QSAR;topomer CoMFA;molecular docking;molecular design;diarylaniline derivatives
2016-03-12;
2016-06-10
国家自然科学基金(21475081);陕西省自然科学基础研究计划(2015JM2057);陕西科技大学研究生创新基金
10.3969/j.issn.1004-4957.2016.11.005
O629.8;TQ460.72
A
1004-4957(2016)11-1397-06
*通讯作者:仝建波,博士,副教授,研究方向:计算机辅助药物设计与相关化学信息与计量学、能源化工、食品化学的研究,Tel:029-86168828,E-mail:jianbotong@aliyun.com
猜你喜欢
含能材料(2022年10期)2022-10-22
吉林大学学报(理学版)(2022年1期)2022-01-21
纺织检测与标准(2021年3期)2021-12-03
大连民族大学学报(2021年5期)2021-11-15
波谱学杂志(2021年3期)2021-09-07
当代化工(2020年9期)2020-11-30
新课程·下旬(2019年7期)2019-09-17
染整技术(2019年2期)2019-04-20
发明与创新·中学生(2017年11期)2017-12-07
中学化学(2015年12期)2016-01-19
