
Natural Bond Orbitals (NBO), Natural Population Analysis,Mulliken Analysis of Atomic Charges of L-Alaninium Oxalate
2016-06-15 16:40CharanyaSampathkrishnanBalamurugan
光谱学与光谱分析 2016年8期
C. Charanya, S. Sampathkrishnan, N. Balamurugan
1. Research Scholar, Department of Physics, Sri Venkateshwara College of Engineering, Sriperumbudur 602105, Tamilnadu, India
2. Department of Physics, Sri Venkateswara College of Engineering, Sriperumbudur 602105, Tamilnadu, India
3. Department of Physics, Sri Lakshmi Ammaal Engineering College, Chennai, Tamilnadu, India
Natural Bond Orbitals (NBO), Natural Population Analysis,Mulliken Analysis of Atomic Charges of L-Alaninium Oxalate
C. Charanya1, S. Sampathkrishnan2, N. Balamurugan3*
1. Research Scholar, Department of Physics, Sri Venkateshwara College of Engineering, Sriperumbudur 602105, Tamilnadu, India
2. Department of Physics, Sri Venkateswara College of Engineering, Sriperumbudur 602105, Tamilnadu, India
3. Department of Physics, Sri Lakshmi Ammaal Engineering College, Chennai, Tamilnadu, India
The Fourier Transform Infrared and Raman spectra of the L-Alaninium oxalate (LAO) have been recorded and analyzed. The fundamental vibrational wave numbers intensities of vibrational bands and optimized geometrical parameters of the compound were evaluated using DFT (B3LYP) method with 6-31+G(d,p) basis set. Natural Bond Orbital (NBO) and Natural Population Analysis (NPA) analysis for the LAO compound was carried out. Mulliken population analyses on atomic charges were also calculated.
LAO; DFT; NBO; NCA
Introduction
L-Alaninium oxalate organic single crystal possess unique opto-electronic properties and its molecules have delocalized electron namely, conjugated electron system exhibit various photo responses such as photoconductive, photo catalytic behavior[1-2]. The title compound is asymmetric carbon atom and non-Centro symmetric space group, which make them optically active. It zwitterionic in nature has large hyperpolarizability and non-linear optical effect[3]. In the recent years, amino acid complexes have received much attention because they proved to be useful in nonlinear optical application. In particular, optically active amino acids display specific features of interest such as molecular chirality, wide transparency range in the visible and UV spectral region and zwitterionic nature of the molecule, which favors crystal hardness[4-5].
Literature survey reveals that so far there is no complete theoretical study for the LAO compound. In this study, The redistribution of Electron Density (ED) in various bonding, antibonding orbitals and E(2) energies have been calculated by Natural Bond Orbital (NBO) analysis to give clear evidence of stabilization originating from the hyper conjugation of various intra- molecular interactions. Moreover, the Mulliken population analyses of the LAO compound have been calculated and the calculated results have been reported. The experimental and theoretical results supported each other, and the calculations are valuable for providing a reliable insight into the vibrational spectra and molecular properties.
1 Computational details
The ab initio quantum mechanical calculations were carried out using Gaussian 09W program package[6], invoking gradient geometry optimization[7]. Initial geometry generated from standard geometrical parameter was minimized without any constraint in the potential energy surface in the standard 6-31+G(d,p) basis set. This geometry was then reoptimized at three parameter hybrid functional (B3) Lee-Yang-Parr (LYP) level using 6-31+G(d,p) basis set. Atomic charges and charge transfer energies were assessed by NBO as implemented in Gaussian 09. GaussView and Chemcraft programs were used for visualization.
2 Results and discussion
2.1 NBO analysis
By using the second-order bond-antibond (donor-acceptor) NBO energetic analysis, insight into the most important delocalizationschemes was obtained. The change in electron density (ED) inthe (σ*, π*) antibonding orbitals and E(2) energies have been calculated by natural bond orbital (NBO) analysis[8]using DFT method to give clear evidence of stabilization originating from various molecular interactions. The hyperconjugative interaction energy was deducted from the second-order perturbation approach Thomson[9].
(1)
whereqiis the donor orbital occupancy,εiandεjare diagonal element, andF(i,j) is the off diagonal NBO Fock matrix element. The largerE(2)value the more intensive is the interaction between electron donors and acceptor, i.e. the more donation tendency from electron donors to electron acceptors and the greater the extent of conjugation of the whole system. Delocalization of electron density between occupied Lewis’s type (bond or lone pair) NBO orbital and formally unoccupied (anti bond or Rydberg) non-Lewis NBO orbital corresponds to a stabilizing donor-acceptor interaction. NBO analysis has been performed on the LAO molecule at the B3LYP/6-31+G(d,p) level to elucidate, the intra-molecular rehybridization and delocalization of electron density within the molecule. The strong intramolecular hyperconjugation interaction of the σ and π electrons of C—C to the anti C—C bond to the ring leads to stabilization of some part in the ring as evident from Table 1 The intra molecular hyperconjugative interaction of the (C1—C3) distribute to (C1—O5), (N2—H19) ,(N2—H21), (C3—C4) and (C4—H24) leading to stabilization of 0.83, 0.059, and 3.57 kJ·mol-1respectively. This enhanced further conjugate with anti bonding orbital ofσ*(C1—C3), (C7—C9), leads to strong delocalization of 4.70 and 1.25 kJ·mol-1respectively.
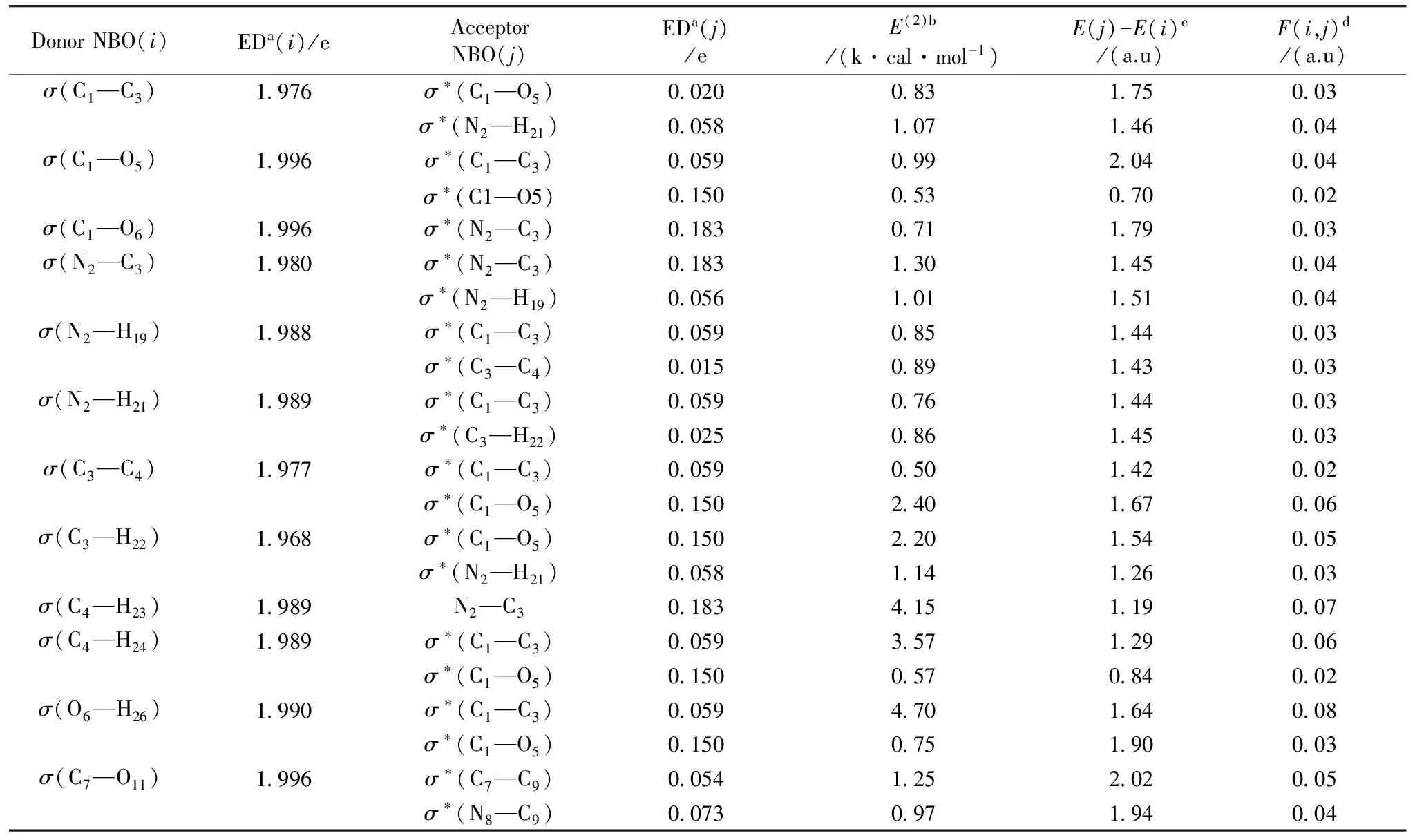
Table 1 Second-order perturbation theory analysis of Fock matrix in NBO
2.2 Natural population analysis
Natural Population Analysis[10](NPA) data of monomer and tetramer were used to investigate the changes in charge, which give some insight into the interaction taking place upon aggregation. For the sake of comparison, the calculated natural charges of LAO are presented in Table 2. It shows that an atoms C13has the most electronegative charge of -5.239 61 and O6has the most electropositive charge of 8.803 48e. Likewise, C1 and C14 atom has considerable electro negativity and they are tending to donate an electron. Conversely, the C3, C4and C9atoms have considerable electropositive and they are tending to acquire electron. Further, the natural population analysis showed that 142 electrons in the LAO molecule are distributed on the sub shell as follows:
Core: 35.989 80 (99.9717% of 36)
Valence: 105.554 77 (99.580 0% of 106)
Rydberg: 0.455 43 (0.320 7% of 142)

Table 2 Accumulation of natural charges and electron population of atoms
a: Atoms containing negative charges;
b: Atoms containing positive charges
2.3 Mulliken population analysis
The Mulliken atomic charges are calculated by determining the electron population of each atom as defined by!the basis function[11]. The Mulliken atomic charges of LAO molecule calculated by B3LYP/6-31+G(d,p) basis set. Calculation of effective atomic charges plays an important role in the application of quantum chemical calculations to molecular systems. Our interest here is in the comparison of different methods to describe the electron distribution in LAO as broadly as possible, and assess the sensitivity, the calculated charges to changes in (1) the choice of the basis set; (2) the choice of the quantum mechanical method. Mulliken charges, calculated the electron population of each atom defined in the basic functions. The Mulliken charges calculated at different levels and at same basis set are listed in Table 3. The results can, however, better represent in graphical form as given Figure 1. The charges depending on basis set are changed due to polarizability. The H20and H21atoms have more positive charges at B3LYP/6-31+G(d,p). This is due to the presence of electronegative oxygen atom; the hydrogen atoms attract the positive charge from the oxygen atoms The C1, C7and C8atoms by B3LYP method is more negative charges than the other atoms due to electron accepting substitutions at that position in LAO. The result suggests that the atoms bonded to O atom and all H atoms are electron acceptor and the charge transfer takes place from O to H in LAO.
Table 3 Mulliken population analysis of L- Alaninium oxalate performed at B3LYP method with 6-31+G (d,p) basis set
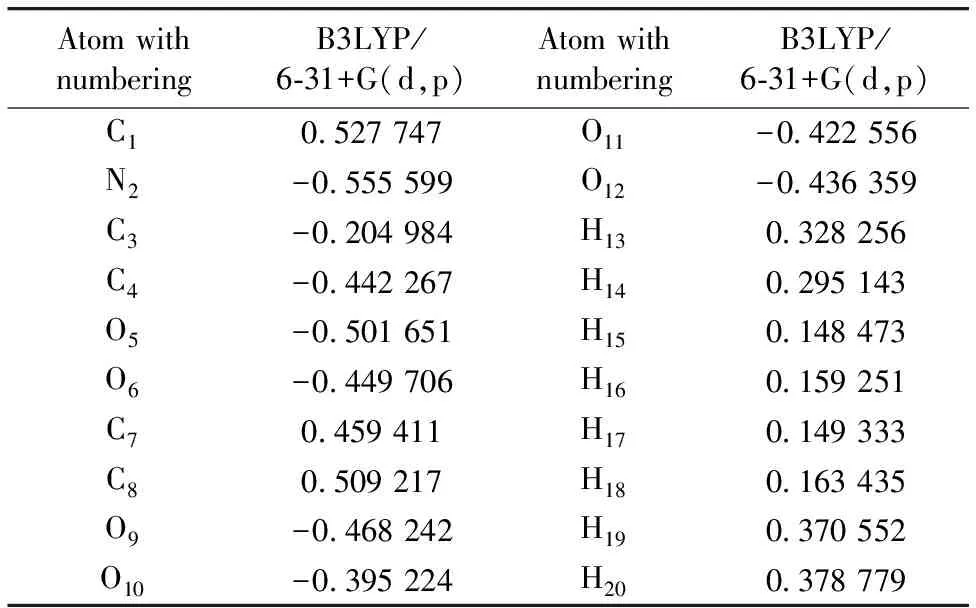
AtomwithnumberingB3LYP/6⁃31+G(d,p)AtomwithnumberingB3LYP/6⁃31+G(d,p)C10 527747O11-0 422556N2-0 555599O12-0 436359C3-0 204984H130 328256C4-0 442267H140 295143O5-0 501651H150 148473O6-0 449706H160 159251C70 459411H170 149333C80 509217H180 163435O9-0 468242H190 370552O10-0 395224H200 378779
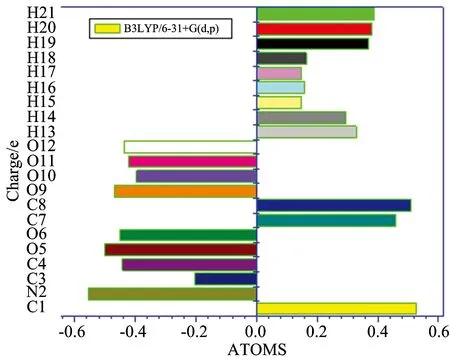
Fig.1 Mulliken population analysis chart
3 Conclusion
In the present work, the optimized molecular structure of the L-Alaninium oxalate compound have been calculated by DFT/B3LYP method with 6-31+G(d,p) level calculations. The NBO analysis provides an efficient method for studying inter and intra molecular interaction in molecular system. The stabilization energy has been calculated from second order perturbation theory. NBO theory provides an excellent approach to interpret the infrared spectra in electronic terms. Relative band positions, together with displacement of bands corresponding to molecular groups not involved in hydrogen bonds, are explained by this theory. In this paper, NBO analysis was also used to evaluate the influence of the intermolecular interactions by comparing the results obtained for the single point and optimized structures. The NBO analysis confirms the hyper conjugation interaction. The strengthening and increase in wave number is due to the substitution of OH and CH3groups, respectively.
[1] Nalwa H S, Miyata S. Non Linear Optics of Organic Molecules and Polymers’. CRC Press, Boca Raton, 1997.
[2] Gunter P. Non Linear Optical Effects And Materials. Springer Verlag, Berlin, 2000.
[3] Moitra S, Kar T. Crystal Research and Technology, 2009, 45: 70.
[4] Rameshbabu R, Vijayan N, Gopalakrishnan R, et al. Crystal Research and Technology, 2006, 41: 405.
[5] Mohankumar R, Rajanbabu D, Jayaraman D, et al. J. Cryst. Growth, 2005, 275: 1935.
[6] Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian 09, Revision A I, Gaussian, Inc, Pittsburgh, P A, 2009.
[7] Schlegel H B. J. Comput. Chem., 1982, 3: 14.
[8] Glendening E D, Reed A E, Carpenter J E, et al. NBO Version 3.1, TCI, University of Wisconsin, Madison, 1998.
[9] Thomson H W, Torkington P. J. Chem. Soc., 1945, 171: 640.
[10] Reed A E, Weinstock R B, Weinhold F. J. Chem. Phys., 1985, 83: 735.
[11] Mulliken R S. J. Chem. Phys., 1995, 23: 1833.
O56
A
2015-08-31; accepted: 2015-12-20
10.3964/j.issn.1000-0593(2016)08-2721-04
*Corresponding author, PhD (spectroscopy), Dhanalakshim College of Engineering, India e-mail: n_rishibalaa@yahoo.co.in
