
Profiling and Validation of the Circular RNA Repertoire in Adult Murine Hearts
2016-11-17 08:41TobiasJakobiLisaCzajaHasseRichardReinhardtChristophDieterichd
Tobias Jakobi*,Lisa F.Czaja-Hasse,Richard Reinhardt,Christoph Dieterichd
1Section of Bioinformatics and Systems Cardiology,Department of Internal Medicine III,University Hospital Heidelberg,69120 Heidelberg,Germany
2German Centre for Cardiovascular Research(DZHK),Heidelberg/Mannheim,Germany
3Max Planck-Genome-Centre Cologne,50829 Cologne,Germany
ORIGINAL RESEARCH
Profiling and Validation of the Circular RNA Repertoire in Adult Murine Hearts
Tobias Jakobi1,2,*,a,Lisa F.Czaja-Hasse3,b,Richard Reinhardt3,c,Christoph Dieterich1,2,d
1Section of Bioinformatics and Systems Cardiology,Department of Internal Medicine III,University Hospital Heidelberg,69120 Heidelberg,Germany
2German Centre for Cardiovascular Research(DZHK),Heidelberg/Mannheim,Germany
3Max Planck-Genome-Centre Cologne,50829 Cologne,Germany
Received 20 November 2015;revised 19 January 2016;accepted 15 February 2016 Available online 27 April 2016
Handled by Andreas Keller
Circular RNA;
circSeq;
Cardiovascular disease;
Cardiomyopathy;
Computational cardiology
For several decades,cardiovascular disease has been the leading cause of death throughout all countries.There is a strong genetic component to many disease subtypes(e.g.,cardiomyopathy)and we are just beginning to understand the relevant genetic factors.Several studies have related RNA splicing to cardiovascular disease and circular RNAs(circRNAs)are an emerging player.circRNAs,which originate through back-splicing events from primary transcripts,are resistant to exonucleases and typically not polyadenylated.Initial functional studies show clear phenotypic outcomes for selected circRNAs.We provide,for the first time,a comprehensive catalogue of RNase R-resistant circRNA species for the adult murine heart.This work combines state-of-the-art circle sequencing with our novel DCC software to explore the circRNA landscape of heart tissue. Overall,we identified 575 circRNA species that pass a beta-binomialtest for enrichment(false discovery rate of 1%)in the exonuclease-treated sequencing sample.Several circRNAs can be directly attributed to host genes that have been previously described as associated with cardiovascular disease.Further studies of these candidate circRNAs may reveal disease-relevant properties or functions of specific circRNAs.
Introduction
Circular RNAs(circRNAs)are a recently-rediscovered RNA class,which were initially described as‘scrambled”exons in the early 1990s[1].Those structures of out-of-order exons may either result from tandem exon duplications,transsplicing,or joining splice donor and acceptor sites of a transcript,thus creating a circular construct.In 2013,severalground-breaking manuscripts demonstrated a clear phenotypic outcome after circRNA perturbation and sparked enormous interest in this molecular species[2-4].Since then,a plethora of manuscripts on circRNA predictions in various contexts has appeared in the literature.circRNAs are typically identified in rRNA-depleted sequencing samples as back-splicing events[5]that break the collinearity between RNA and DNA sequences.However,this necessary condition is not sufficient to prove circularity of the RNA species,since‘scrambled exons”may also arise from trans-splicing and tandem duplications as mentioned above,genomic alterations,or simply from read mapping artefacts[6].
The identification and quantification of circRNAs from next-generation sequencing(NGS)data is an area of ongoing research.The‘circSeq”approach is the current gold standard for high-throughput circRNA identification[7].Briefly,circRNAs are resistant to exonuclease treatment,whereas linear RNAs are selectively depleted with this approach.An untreated control sample and an RNase R-treated sample are subjected to RNA-seq and contrasted with one another. Although circSeq is the current gold standard,it may not work equally well on all circRNAs.For instance,CDR1as/ciRS-7 could not be enriched with RNase R treatment[8].
Relative abundance changes in circRNA expression are typically approximated by comparing normalized back-splice junction read counts of specific circRNAs under different conditions and/or time points[9].Moreover,we recently presented a statistical framework to detect host gene-independent circRNA expression changes from sequencing data[10].
It is widely assumed that circRNAs belong to the growing group of non-coding RNAs(ncRNAs)that encompasses microRNAs(miRNAs),long ncRNAs(lncRNA),or smallinterfering RNAs(siRNA).Intriguingly,a recent study in endothelial cells showed that circRNAs are regulated in response to hypoxia and have a biological function as proven by in vitro experiments[11].Nonetheless,no rigorous reports have been published on circRNAs in murine or human heart cell types.An extrapolation from other tissue or cell type data is not possible as previous analyses have clearly demonstrated that a large proportion of circRNAs are cell type specific[12].
In this manuscript,we aim to provide a catalogue of validated circRNA species in the adult murine heart.To this end,we employ the latest developments in bioinformatics and sequencing technology,and relate our catalogue to previously-identified cardiomyopathy-related genetic loci.
Results and discussion
Total RNA was extracted from hearts of adult mice which were 2,3,6,or 12 months old.After depletion of rRNA,each RNA sample was split into two fractions with one treated with RNase R(RNase R+)and the other left untreated(RNase R-)(see Methods).No polyA-containing RNA removal step was carried out,in accordance with other recent reports on fragment-based circRNA detection[7]and due to the fact that otherwise no comparison of linear transcript counts to circular counts would be possible.We prepared cDNA libraries from these 8 samples and sequenced them on an Illumina HiSeq 2500.The corresponding read coverage tracks have been compiled into a publicly available TrackHub for the UCSC Genome Browser[13](Supplementary online data).

Table1 Selection of 8 significant circRNAs linked to certain types of cardiovascular disease
575 circRNAs are significantly enriched
The DCC software[10]was used to identify circRNA candidates from chimeric read mappings(see Materials and Methods).This initial step yielded 8375 potential circRNA regions.We subsequently employed the circTest suite in DCC[10]to confine the search to 741 circRNA lociby filtering for consistent read support and a minimal proportion of junction-spanning reads(1%).Lastly,we tested enrichment of back-splicing in read sets of RNase R+in comparison to RNase R-samples and further reduced the candidate circRNA set to 575 circRNAs(Table S1).We additionally screened the catalogue of the 575 significant circRNAs for alternative splicing using the DCC output files and found only 2.4%having>3 supporting reads for alternative splicing variants.
Table 1 shows an example of 8 selected circRNAs that are localized in cardiovascular disease-related gene loci.circTest generates enrichment plots for each of the significant circRNAs(BH corrected P values,FDR 1%).Due to the employed circle sequencing approach,RNase R-and RNase R+samples should be clearly separated within the enrichment plot since the RNase R digestion step removes the bulk of linear transcripts and more sequencing reads actually cover circRNAs structures instead of linear transcripts in the untreated sample.Therefore,this graphical representation is a first quality measurement for the success of enrichmentprocess.In an ideal experiment all linear fragments in the RNase Rtreated sample should be digested,leaving only circular constructs in the sample.A drastic increase of reads covering circRNAs would therefore be expected when comparing untreated and treated samples.Also this ideal situation does not translate exactly to practice.The enrichment plots in Figure 1 show that the enrichment process performed well for all samples,thus confirming the gold standard of circle sequencing.
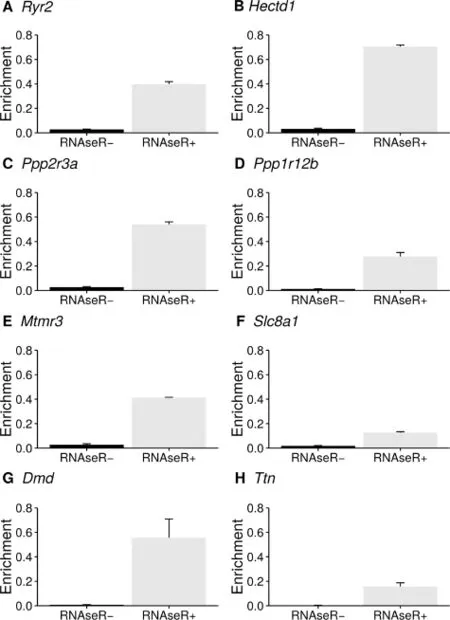
Figure1 Comparison of the enrichment for circRNAs in RNase R+and RNase R-samples
Inspection of the most significant circRNAs
The vast majority of circRNAs were only found at only one location within the annotated gene regions(97.6%,threshold: 3 supporting reads).For the remaining 2.4%circRNAs,more than 3 reads were found at a secondary location within the annotated gene regions;however,hardly more than 10 reads were detected at the secondary locations for these circRNAs. Therefore,circRNAs seem to be predominantly emerging fromsingle,specific gene regions.Among the 575 significant circRNAs identified above,we selected the top three most significant loci,Ryr2,Hectd1,and Ppp2r3a that encode ryanodine receptor 2,HECT domain-containing E3 ubiquitin protein ligase 1,and protein phosphatase 2,regulatory subunit B,respectively,for further inspection.
The annotated genomic region of Ryr2,throughout all experiments,showed the most significant enrichment in general(Table 1,Figure 1).The combination of expression level,enrichment compared to the remaining linear RNAs reported by circTest(Figure 2),and its involvement in arrhythmias make Ryr2 circRNA a very promising candidate for further analyses.The circRNA for Ryr2 is roughly located between the second and last third of the annotated gene region(Figure 3).The RNase R-circRNA candidate reads cover a region of 6 exons and 5 introns,starting at exon 24 and up to exon 29 of 25 kb in length.

Figure2 Detailed view of five selected genes strongly related to cardiovascular disease

Figure3 Ensemblscreen shot of the circRNA locus linked to the Ryr2 gene
The locus of the Hectd1 circRNA spans only 432 bp.The fragment spans only 2 exons(namely exon 23 and exon 24)and is located within the last third of Hectd1 coding region(Figure 4).Hectd1 circRNA belongs to the class of relatively small circRNAs.Notwithstanding,it is one of the most significant circRNAs in our samples.
In contrast,the identified circRNA locus of Ppp2r3a again measures more than 28 kb and contains 6 exons,starting from exon 6 and ranging up to exon 12.Due to the small size of Ppp2r3a,the 28 kb circRNA makes up nearly a quarter of the gene’s length and is located in the last quarter of the annotated gene region(Figure 5).

Figure4 Ensembl screen shot of the circRNA locus linked to the Hectd1 gene

Figure5 Ensembl screen shot of the circular RNA locus linked to the Ppp2r3a gene
Identification of candidate cardiovascular circRNAs
circRNAs are known to be highly specific to tissue and developmental status of the cell[12].An in-depth analysis of highlyabundant circRNAs disclosed several genes tightly linked to functions of cardiomyocytes.Due to the selection of heart tissue and our focus on cardiovascular disease,we focused on three genes tightly coupled to cardiomyocytes and cardiomyopathy,Ryr2,Ttn,and Dmd for scrutinization.
These candidate genes are known to play key roles in different kinds of cardiomyopathy such as Dmd-associated dilated cardiomyopathy,arrhythmogenic right ventricular cardiomyopathy(ARVC)(Ryr2),or hypertrophic cardiomyopathy(HCM,Ttn),and therefore are central parts of the respective KEGG pathways(mmu05414,mmu05410,and mmu05412)[22].
There is an overwhelming presence of Ryr2 circRNAs regardless ofthe age ofthe murine heartexamined(Figure 2A). Primarily expressed in cardiomyocytes,Ryr2 encodes ryanodine receptor 2(RYR2),which is a sarcoplasmic reticulum membrane-embedded transport protein for Ca2+and is responsible for the rhythmic muscle contractions generating the heartbeat[14].The abundance of Ryr2 circRNAs is relatively constant for all 4 time points,which peaks at month 6. Traces of Ryr2 circRNA are detectable even without the enrichment step,whereas after enrichment the abundance is 2-3 times higher when compared to the untreated sample.
The detection of Ttn fits in the same picture,as it is responsible for the passive elasticity of muscles and therefore plays a critical role in heart muscle cells[21].circRNA abundance of Ttn generally follows the pattern of Ryr2 albeit with relatively modest changes.In comparison,the enrichment process was not as successful as for Ryr2,as higher circRNA counts were detected for month 4 and month 6 in the untreated samples than the treated samples.For example,very long circRNA molecules may get nicked or sheared during the isolation process,making them sensitive to RNAse R treatment.
Dmd-associated dilated cardiomyopathy is directly linked to mutations in the Dmd gene[20].Therefore,Dmd circRNA could be another valuable target for cardiomyopathy research. The expression pattern of Dmd circRNA differs from those of Ryr2 and Ttn,demonstrating maximal expression for month 12 instead of month 6.We achieved non-optimal enrichment for circRNAs from mice aged 3 or 6 months,possibly due to portions of the circRNAs lost during the RNase R digestion process.
The pattern of abundance of circRNAs for all three genes may be influenced by the age and activity of the mice.While the linear transcripts Ryr2 and Ttn may be more connected to young and active hearts,Dmd-encoded protein strengthens the cardiomyocyte sarcolemmal integrity,and its maximal expression may be important for optimal function(hemodynamic and stress responses)ofthe fully developed,mature adult heart. Although the described properties are related to the proteincoding linear transcripts,their corresponding circRNAs may be involved in those diseases directly or indirectly.
Conclusion
In this work we present circle sequencing as a highly efficient and sensitive tool for circRNA studies in cardiology.In summary,we compiled a catalogue of 575 candidate circRNAsthat pass a test for enrichment in RNase R-treated samples in comparison to untreated samples using circTest.Many of these candidates coincide with disease-associated gene loci.For instance,significant candidates originate from the Ryr2,Hectd1,and Ppp2r3a gene loci that have been linked to cardiovascular disease.
As a long term goal,how these circRNAs might influence cell processes in disease-related phenotypes and healthy tissue needs to be addressed.Furthermore,more efforts are also needed as to how this information may translate into new treatment approaches of cardiovascular disease.
Materials and methods
Circle sequencing
Total RNA extracted from heart tissue of CD1-strain mice of ages 2,3,6,and 12 months was commercially acquired from AMS Biotechnology(Abingdon,Oxfordshire,UK).The rRNA depletion step was carried out with 20μg of total RNA using the Ribo-Zero Magnetic Kit(Human,Mouse,Rat;Epicentre,Madison,WI,United States)and 5μg input per reaction.Depletion was performed in parallel,followed by subsequent pooling.For digestion of linear RNA,3 quarters of the depleted RNA was treated with RNase R,whereas the remaining one quarter of the depleted RNA only received water and acts as a negative control.Both samples were treated equally for all further steps.After heating to 70°C and cooling to 35°C,10×RNase R reaction buffer and RNase R(Epicentre;Cat No.RNR07250)were added to the 3-quarter fraction,followed by heating to 37°C for 40 min.Samples were cleaned up using Agencourt RNAClean XP beads(Beckman Coulter GmbH,Krefeld,North Rhine-Westphalia,Germany).Subsequent library preparation was performed with ScriptSeq-v2 RNA-Seq Library Preparation Kit(Epicentre).PCR amplification was performed with 15 cycles,without any further size selection.Sequencing was carried out on an Illumina HiSeq2500 system for 100 cycles using paired-end mode. Sequence data were deposited in the NCBI Sequence Read Archive(SRA)with the accession number SRP071584.
Bioinformatics analyses
Paired-end rRNA-depleted sequencing data of RNase R treated and untreated samples were analysed in detail.After initial quality assessment,low quality regions and adapter sequences were removed with Flexbar(version 2.5)[23].Read mapping against the mouse reference genome build mm10 was performed with the STAR aligning software(version 2.4.2a)[24].
To optimally exploit the advantage of the circSeq technique,we have developed the DCC software to efficiently detect and quantify circRNAs in sequencing data[10].DCC software computes expression levels of host genes and circRNAs and allows for tests of circRNA expression independent of linear host genes even across several experimental conditions and replicates.DCC analysis took place with version 0.3.2 using the parameters‘-M-Nr 2 2-fg-G-F-L 20”.Further statistical analyses were performed with the circTest R package provided with DCC and additional custom-tailored Perl scripts.
Authors’contributions
CD designed the study;LC and RR performed all RNA and sequencing experiments.TJ and CD analysed the data and wrote the manuscript.All authors read and approved the final manuscript.
Competing interests
The authors declared that there are no competing interests.
Acknowledgments
This work was supported by the Klaus Tschira Stiftung,Heidelberg,Germany.The authors would like to thank the Max Planck Institute for Biology of Ageing,Cologne and especially the Bioinformatics Core Facility for providing access to the high performance compute cluster.
Supplementary material
The TrackHub containing data of this study in bigWig format is available at https://trackhub.dieterichlab.org/tjakobi/ circRNA_heart/hub.txt and a preconfigured instance is accessible via http://tiny.cc/circRNA.Supplementary material associated with this article can be found,in the online version,at http://dx.doi.org/10.1016/j.gpb.2016.02.003.
[1]Nigro JM,Cho KR,Fearon ER,Kern SE,Ruppert JM,Oliner JD,et al.Scrambled exons.Cell 1991;64:607-13.
[2]Hansen TB,Jensen TI,Clausen BH,Bramsen JB,Finsen B,Damgaard CK,et al.Natural RNA circles function as efficient microRNA sponges.Nature 2013;495:384-8.
[3]Memczak S,Jens M,Elefsinioti A,Torti F,Krueger J,Rybak A,et al.Circular RNAs are a large class of animal RNAs with regulatory potency.Nature 2013;495:333-8.
[4]Li Z,Huang C,Bao C,Chen L,Lin M,Wang X,et al.Exonintron circular RNAs regulate transcription in the nucleus.Nat Struct Mol Biol 2015;22:256-64.
[5]Jeck WR,Sharpless NE.Detecting and characterizing circular RNAs.Nat Biotechnol2014;32:453-61.
[6]Lasda E,Parker R.Circular RNAs:diversity of form and function.RNA 2014:1829-42.
[7]Jeck WR,Sorrentino JA,Wang K,Slevin MK,Burd CE,Liu J,et al.Circular RNAs are abundant,conserved,and associated with ALU repeats.RNA 2013;19:141-57.
[8]Chen I,Chen CY,Chuang TJ.Biogenesis,identification,and function of exonic circular RNAs.Wiley Interdiscip Rev RNA 2015;6:563-79.
[9]Ivanov A,Memczak S,Wyler E,Torti F,Porath HT,Orejuela MR,et al.Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals.Cell Rep 2015;10:170-7.
[10]Cheng J,Metge F,Dieterich C.Specific identification and quantification of circular RNAs from sequencing data.Bioinformatics 2016;32:1094-6.
[11]BoeckelJN,Jae´N,Heumu¨ller AW,Chen W,Boon RA,Stellos K,et al.Identification and characterization of hypoxia-regulated endothelial circular RNA.Circ Res 2015;117:884-90.
[12]Salzman J,Chen RE,Olsen MN,Wang PL,Brown PO.Cell-type specific features of circular RNA expression.PLoS Genet 2013;9: e1003777.
[13]Kent WJ,Sugnet CW,Furey TS,Roskin KM,Pringle TH,Zahler AM,et al.The human genome browser at UCSC.Genome Res 2002;12:996-1006.
[14]Wellcome Trust Case Control Consortium.Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls.Nature 2007;447:661-78.
[15]Anderson E,Khvorova A,Seto AG,Baskerville S,Leinwand L,Sullivan KG.Identification of miRNA profiles that are diagnostic of hypertrophic cardiomyopathy.2010;US Patent US20110160285 A1.
[16]Zhou X,Song G,Gan X,Li C,Yang N,Zheng F.Human PPP2R3A genetic mutation and application thereof.2012;China Patent CN102443630A.
[17]Domarkiene I,Pranculis A,Germanas Sˇ,Jakaitiene A,Vitkus D,Dzˇenkevicˇiute V,et al.Rtn4 and Fbxl17 genes are associated with coronary heart disease in genome-wide association analysis of Lithuanian families.Balk J Med Genet 2013;16:17-22.
[18]Willer CJ,Schmidt EM,Sengupta S,Peloso GM,Gustafsson S,Kanoni S,et al.Discovery and refinement of lociassociated with lipid levels.Nat Genet 2013;45:1274-83.
[19]Deng X,Sabino EC,Cunha-Neto E,Ribeiro AL,Ianni B,Mady C,et al.Genome wide association study(GWAS)of Chagas cardiomyopathy in Trypanosoma cruzi seropositive subjects.PLoS One 2013;8:e79629.
[20]Muntoni F,Torelli S,Ferlini A.Dystrophin and mutations:one gene,several proteins,multiple phenotypes.Lancet Neurol 2003;2:731-40.
[21]Herman DS,Lam L,Taylor MR,Wang L,Teekakirikul P,Christodoulou D,et al.Truncations of titin causing dilated cardiomyopathy.N Engl J Med 2012;366:619-28.
[22]Kanehisa M,Goto S.KEGG:Kyoto encyclopedia of genes and genomes.Nucleic Acids Res 2000;28:27-30.
[23]Dodt M,Roehr JT,Ahmed R,Dieterich C.FLEXBAR—flexible barcode and adapter processing for next-generation sequencing platforms.Biology(Basel)2012;1:895-905.
[24]Dobin A,Davis CA,Schlesinger F,Drenkow J,Zaleski C,Jha S,et al.STAR:ultrafast universal RNA-seq aligner.Bioinformatics 2013;29:15-21.
*Corresponding author.
E-mail:tobias.jakobi@med.uni-heidelberg.de(Jakobi T).
aORCID:0000-0002-3906-0401.
bORCID:0000-0002-3261-4424.
cORCID:0000-0001-9376-2132.
dORCID:0000-0001-9468-6311.
Peer review under responsibility of Beijing Institute of Genomics,Chinese Academy of Sciences and Genetics Society of China.
http://dx.doi.org/10.1016/j.gpb.2016.02.003
1672-0229©2016 The Authors.Production and hosting by Elsevier B.V.on behalf of Beijing Institute of Genomics,Chinese Academy of Sciences and Genetics Society of China.
This is an open access article under the CC BY license(http://creativecommons.org/licenses/by/4.0/).
 Genomics,Proteomics & Bioinformatics2016年4期
Genomics,Proteomics & Bioinformatics2016年4期
- Genomics,Proteomics & Bioinformatics的其它文章
- A Biobank for Long-term and Sustainable Research in the Field of Congenital Heart Disease in Germany
- Long non-coding RNA Databases in Cardiovascular Research
- The Role of Quality Control in Targeted Next-generation Sequencing Library Preparation
- Comparative Gene Expression Analysis of Mouse and Human Cardiac Maturation
- Absent MicroRNAs in Different Tissues of Patients with Acquired Cardiomyopathy
- Comparison of Cox Model Methods in A Low-dimensional Setting with Few Events