
基于α螺旋肽与多酚缀合的HIV-1融合抑制剂的设计、 合成及活性初筛
2016-12-27 02:12程思绮梁国栋姜喜凤刘克良
高等学校化学学报 2016年7期
程思绮, 梁国栋, 姜喜凤, 王 潮, 刘克良,
(1. 沈阳药科大学制药工程学院, 沈阳 110016;2. 军事医学科学院毒物药物研究所, 北京 100850)
基于α螺旋肽与多酚缀合的HIV-1融合抑制剂的设计、 合成及活性初筛
程思绮1, 梁国栋2, 姜喜凤2, 王 潮2, 刘克良1,2
(1. 沈阳药科大学制药工程学院, 沈阳 110016;2. 军事医学科学院毒物药物研究所, 北京 100850)
将α螺旋多肽与羟基酪醇等多酚类化合物通过共价键缀合, 期望二者在发挥各自生物学作用的同时产生协同效应, 据此设计了HIV-1融合抑制多肽. 圆二色光谱表征结果表明, 所设计的缀合多肽呈典型的α螺旋结构特征, 且可以与靶标N36相互作用. HIV-1包膜糖蛋白(Env)介导的细胞-细胞融合活性测试结果表明, 这些具有α螺旋结构的缀合多肽可以在低微摩尔水平抑制病毒融合.
人免疫缺陷病毒Ⅰ型; 融合抑制剂;α螺旋肽; 多酚
人免疫缺陷病毒Ⅰ型(Human Immunodificiency virus type 1, HIV-1)侵染宿主细胞的过程由病毒包膜糖蛋白(Envelope glycoproteins, Env)介导. 包膜糖蛋白跨膜亚基gp41中N末端重复序列(N-heptad repeat, NHR)和C末端重复序列(C-heptad repeat, CHR)可发生相互作用, 形成六股α螺旋束(Six helix bundle, 6HB), 这是病毒膜和细胞膜融合的关键步骤[1,2]. 在6HB的形成过程中, 3股NHR首先形成三聚体内核(N-trimer), 3股CHR再以反向平行的方式结合到N-trimer形成的疏水性沟槽中[3]. 一些衍生于病毒CHR区域的多肽可以与靶标N-trimer作用, 干预内源性6HB的生成, 从而阻断膜融合过程, 抑制病毒进入宿主细胞[4,5]. 这类CHR区域衍生多肽称为C肽类HIV-1融合抑制剂[6~9]. 其中, T20(商品名Fuzeon, 通用名Enfuvirtide)是衍生于HIV-1 gp41 CHR的36肽, 已于2003年由美国食品药品管理局(FDA)批准上市[10].
由C34/N36所形成6HB的晶体结构可知, 6HB本质为α螺旋间彼此相互作用形成的卷曲螺旋(Coiled coil)结构[11]. 在该超螺旋结构中, 每隔7个氨基酸残基, 单股α螺旋正好转过2圈, 其位置结构可表示为(abcdefg)n. 各位置上的残基分布具有如下规律: (1) a, d位通常为以亮氨酸(Leu)为代表的疏水性残基, 而e, g位通常为极性残基; (2) NHR首先依靠a, d位残基的疏水相互作用形成N-trimer, 进一步通过其表面的e, g位残基与CHR中的a, d位残基相互作用形成6HB[如图1(A)和(B)所示]. 6HB晶体结构分析结果表明, C肽的活性构象为亮氨酸拉链样的α螺旋. 适度提高天然C肽的α螺旋含量, 可有效提高药物的抗病毒活性[12,13]. 前文[14]曾从头设计了一种具有重复拷贝的4-3七残基序列5HR, 即(AEELAKK)5. 生物物理学及生物学活性评价表明, 5HR在溶液中可形成稳定的α螺旋结构, 且具有一定的HIV-1融合抑制活性. Shi等[15]以5HR为先导结构, 在其N端引入衍生于HIV-1 CHR的寡肽序列PBD(aa: 628~634, WMEWDRE), 同时对螺旋a, d位残基进行定点突变, 得到了螺旋多肽PBD-m4HR(WMEWEDRE IEELIKK SEELIKK IEEQIKK QEESIKK). 该序列活性与天然C肽相当, 且能与靶标NHR形成稳定的6HB. 由5HR及PBD-m4HR的发现可见, 在6HB形成过程中非特异性的疏水作用和静电作用的贡献很大, 残基一一对应的特异性结合序列有可能被形成α螺旋的多肽序列所替代.
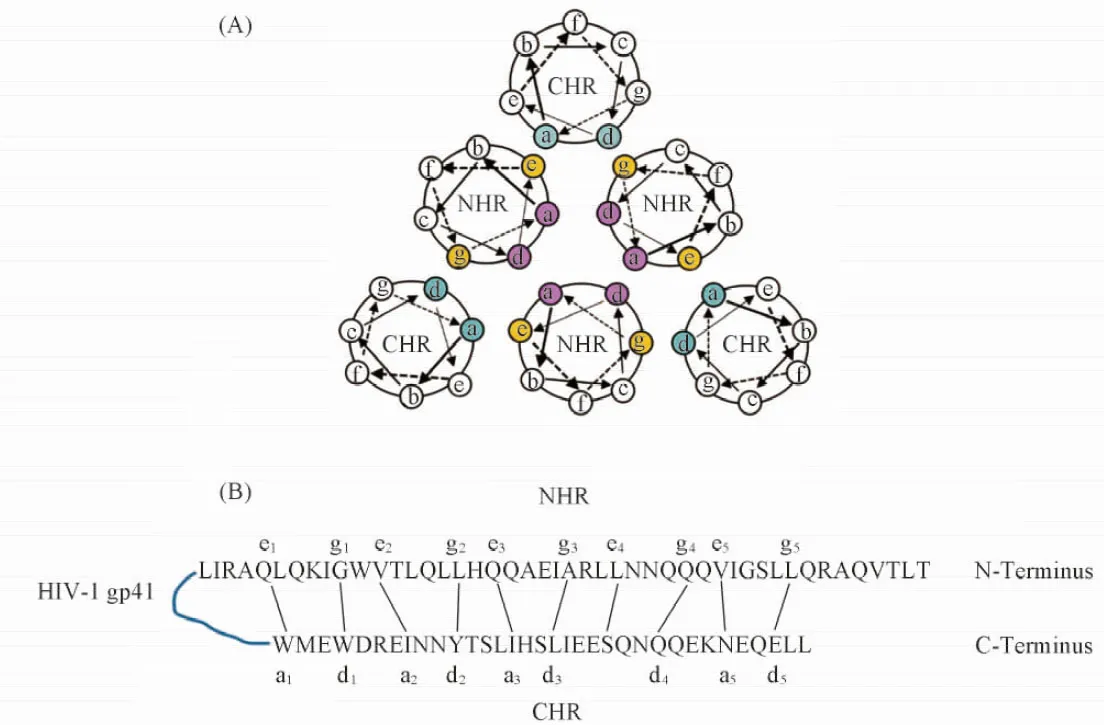
Fig.1 Ineraction between NHR and CHR of HIV gp41
多酚是一类重要的天然产物, 具有抗病毒、 抗肿瘤及抗炎等多种生物学活性[16~19]. 多酚的抗病毒活性具有广谱性, 如表没食子儿茶素没食子酸酯(Epigallocatechin gallate, EGCG)能够同时阻止HIV-1和流感病毒进入宿主细胞[20]. 此外, 多酚亦可作用于某一病毒生命周期的多个环节, 如在阻止HIV-1侵染宿主细胞的过程中, 多酚不仅能够通过与逆转录酶及多种蛋白酶结合抑制病毒复制过程, 还能作用于病毒的进入阶段[21,22]. 上述多酚与靶标之间的非特异性结合特点导致其与靶标的作用力不强、 生物活性不高. 前期研究[23]表明, 从橄榄叶中提取的羟基酪醇(HT)和橄榄苦苷(Ole)能够阻断HIV-1与宿主细胞的融合过程, 发挥抗病毒活性. HT的HIV-1融合抑制机制主要是与gp41中的N-trimer结合, 从而抑制内源性6HB的生成.
本文基于6HB的超螺旋结构特征, 将用于靶向病毒NHR的α螺旋多肽与具有非特异性抗病毒作用的多酚类化合物缀合. 在所设计的缀合物中,α螺旋肽具有NHR靶向性, 可以将多酚有效带至靶标部位, 从而最大程度地释放出多酚的抗病毒融合活性; 同时, 多酚的存在也势必会影响α螺旋肽的高级结构与理化性质. 活性多酚和α螺旋肽在结构和功能上互相影响、 互为补充, 可能会取得1+1>2的协同效应, 得到具有HIV-1融合抑制活性的新颖结构. 我们从PBD-m4HR中截取了m4HR部分[24], 并通过不同长度和柔性的连接臂将其与HT衍生物mHT或其它具有类似结构的多酚缀合, 以考察α螺旋肽与多酚的协同作用.
1 实验部分
1.1 试剂与仪器
Rink-amide树脂(载量0.44 mmol/g)购于天津南开和成科技有限公司; 三氟乙酸(TFA)购于北京博迈杰科技有限公司; 1,2-乙二硫醇(EDT)、 苯甲醚(Anisole)和间甲酚(m-Cresol)均购于百灵威科技有限公司.
各种保护氨基酸:N-芴甲氧羰基-β-丙氨酸(Fmoc-βAla-OH)、N-芴甲氧羰基-N′-三苯甲基-L-谷氨酰胺[Fmoc-L-Gln(Trt)-OH]、N-芴甲氧羰基-O-叔丁基-L-谷氨酸[Fmoc-L-Glu(OtBu)-OH]、N-芴甲氧羰基-L-异亮氨酸(Fmoc-L-Ile-OH)、N-芴甲氧羰基-L-亮氨酸(Fmoc-L-Leu-OH)、N-α-芴甲氧羰基-N′-叔丁氧羰基-L-赖氨酸[Fmoc-L-Lys(Boc)-OH]、N-芴甲氧羰基-O-叔丁基-L-丝氨酸[Fmoc-L-Ser(t-Bu)-OH]及N-芴甲氧羰基-6-氨基己酸(Fmoc-6-aminocaproic acid)均购自上海吉尔生化有限公司;N-芴甲氧羰基-8-氨基-3,6-二氧杂辛酸(Fmoc-8-amino-3,6-dioxaoctanoic acid)购自浙江嘉兴博美生物有限公司; 原儿茶酸(Protocatechuic acid, PA)购自百灵威公司; 没食子酸(Gallic acid, GA)购自国药集团; 其它所用试剂均为分析纯.
CS Bio Co.多肽合成仪(美国CS公司); Shimadzu LC-20AP型高压制备液相色谱仪(日本岛津株式会社); Phenomenex C8制备液相色谱柱(250 mm×21.20 mm, 美国Phenomenex公司); Shimadzu 10A型高效液相色谱仪(日本岛津株式会社); Waters XBridge C8液相色谱柱(250 mm×4.6 mm, 5 μm, 美国Waters公司); Milli-Q 纯水机(美国MilliPore公司); Free-Zone 18 L 型冷冻干燥机(美国Labconco公司); REFLEX Ⅲ型MALDI-TOF-MS质谱仪(德国Bruker公司).
1.2 化合物5的合成及表征

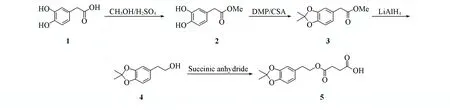
Scheme 1 Synthesis routes of compound 5
1.3 肽序列的合成
采用标准的芴甲氧羰基(Fmoc)固相合成策略, 在CS Bio Co.多肽合成仪上固相合成m4HR. 在m4HR基础上, 同样采取标准的Fmoc固相合成策略(以DIC为缩合剂), 手动连接上不同的连接臂β-丙氨酸(β-Alanine,βAla)、 6-氨基己酸(6-Aminocaproic acid, Aca)和8-氨基-3,6-二氧杂辛酸(8-Amino-3,6-dioxaoctanoic acid, PEG3), 再连接不同的多酚化合物, mHT, PA, GA多酚及缀合物结构如图2所示. 各肽序列均经MALDI-TOF-MS 确证分子量, 利用C8制备型反相色谱柱分离纯化, 得到纯度>95%的纯肽.
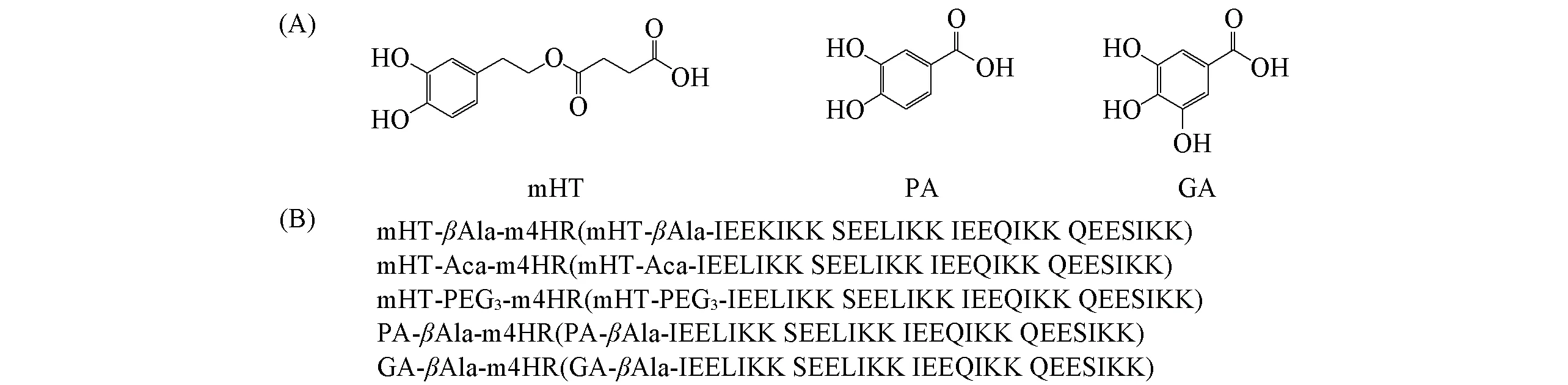
Fig.2 Structures of polyphenols and designed conjugates
1.4 缀合物与N36相互作用的圆二色光谱(CD)测定
配制mHT-βAla-m4HR的PBS(pH=7.4)溶液(10, 20 μmol/L)及N36的水溶液(10, 20 μmol/L). 将mHT-βAla-m4HR溶液(20 μmol/L)及N36溶液(20 μmol/L)各300 μL混合均匀, 在37 ℃下孵育30 min, 得混合溶液, 终浓度为10 μmol/L. 分别测定mHT-βAla-m4HR溶液, N36深液及混合溶液在10 μmol/L浓度水平的CD值.
1.5 HIV-1 Env介导的细胞-细胞融合活性测试
将稀释后的样品与含荧光素酶报告基因的TZM-b1细胞(50×104Cell/mL)及HL2/3细胞(100×104Cell/mL)共孵育24 h后, 裂解细胞, 检测荧光素酶活性(Luciferase assay system). 对样品剂量-融合抑制百分率曲线进行S形拟合(Origin 7.5软件), 得到测试样品的半抑制浓度(IC50)值.
2 结果与讨论
2.1 缀合物与靶序列N36的相互作用
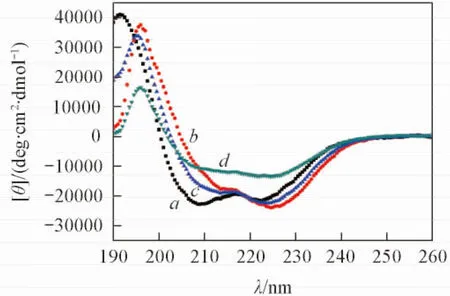
Fig.3 CD spectra of mHT-βAla-m4HR and its complex with gp41 NHR peptides
缀合物与靶序列N36相互作用的CD表征结果如图3所示, 根据各肽序列在222 nm处的CD值, 利用公式:α螺旋度=CD值/33000计算出对应的α螺旋度. mHT-βAla-m4HR呈现典型的α螺旋结构特征, 其自身α螺旋度达到了68.7%. 此结果表明, 缀合物保持了α螺旋活性构象. 此外, mHT-βAla-m4HR与靶序列N36混合孵育后,α螺旋度实测值为40.4%, 而两者α螺旋度的理论加和值为67%. 实测值与理论加和值存在显著差异, 这一结果表明, mHT-βAla-m4HR与靶序列N36产生了一定程度的相互作用[27,28], 破坏了N36所形成的3股α螺旋.
2.2α螺旋肽与多酚之间产生协同效应
HIV-1 Env介导的细胞融合活性测试结果如表1所示, m4HR的活性为36.91 μmol/L. 将多酚类化合物缀合到m4HR的N端后, 缀合物的活性为5.34~22.37 μmol/L, 比m4HR提高了2~7倍. 该结果表明, m4HR和多酚之间产生了协同效应.
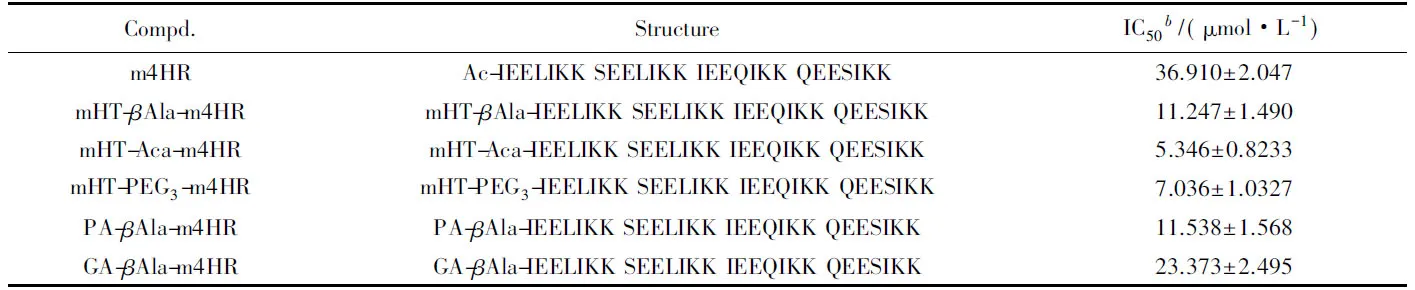
Table 1 Inhibitory activities of conjugates on HIV-1 Env-mediated cell-cell fusiona
此外, 本文还考察了不同类型的多酚及连接臂对协同效应的影响. 在m4HR和mHT之间, 选用了3种不同柔性和长度的连接臂βAla, Aca及PEG3. 其中,βAla结构最简单, 灵活性最弱; PEG3与Aca长度相近, 因结构中存在氧原子而具有更好的柔性. 通过比较mHT-βAla-m4HR, mHT-Aca-m4HR以及mHT-PEG3-m4HR的HIV-1抑制作用发现, 连接臂的类型对缀合物的活性无显著影响. mHT, GA和PA这3种不同的多酚均以βAla为连接臂将其缀合到m4HR上, 分别得到mHT-βAla-m4HR, PA-βAla-m4HR及GA-βAla-m4HR, HIV-1 Env介导的细胞-细胞融合活性测试表明, 三者活性相当, 说明多酚的种类对协同效应无显著影响.
3 结 论
将具有α螺旋的肽序列m4HR与多酚类化合物mHT等通过共价键连接, 得到了仍保持α螺旋活性构象的缀合物. 这些缀合物可与靶标N36作用, 且可以在低微摩尔水平抑制HIV-1融合. 由于含Ⅰ型融合蛋白的包膜病毒具有相似的6HB过渡态结构, 本文结果对包括HIV-1在内的Ⅰ型包膜病毒融合抑制剂的设计具有指导意义.
参 考 文 献
[1] Lu M., Blacklow S. C., Kim P. S.,Nat.Struct.Biol., 1995, 2, 1075—1082
[2] Moore J. P., Doms R. W.,Proc.Natl.Acad.Sci.USA, 2003, 100, 10598—10602
[3] Liu S., Zhao Q., Jiang S.,Peptides, 2003, 24(9), 1303—1313
[4] Donegan E., Stuart M., Niland J. C.,Ann.Intern.Med., 1990, 113(10), 733—739
[5] Cai L. F., Jiang S. B.,Chem.Med.Chem., 2010, 5(11), 1813—1824
[6] Jiang S., Lin K., Strick N., Neurath A. R.,Nature, 1993, 365, 113—114
[7] Wild C. T., Shugars D. C., Greenwell T. K., McDanal C. B., Matthews T. J.,Proc.Natl.Acad.Sci.USA, 1994, 91, 9770—9774
[8] Chen C. H., Greenberg M. L., Bolognesi D. P., Matthews T. J.,AIDSRes.Hum.Retroviruses, 2000, 16(8), 2037—2041
[9] Zhang S., Shi W. G., Wang C., Cai L. F., Zheng B. H., Wang K., Feng S. L., Jia Q.Y., Liu K. L.,Chem.J.ChineseUniversities, 2014, 35(12), 2542—2546(张沙, 史卫国, 王潮, 蔡利峰, 郑保华, 王昆, 冯思良, 贾启燕, 刘克良. 高等学校化学学报, 2014, 35(12), 2542—2546)
[10] Joly V., Jidar K., Tatay M., Yeni P.,ExpertOpin.Pharmacother., 2010, 11, 2701—2713
[11] Chan D. C., Fass D., Berger J. M., Kim P. S.,Cell, 1997, 89(2), 263—273
[12] Judice J. K., Tom J. Y., Huang W., Wrin T., Vennari J., Petropoulos C. J., McDowell R. S.,Proc.Natl.Acad.Sci.USA, 1997, 94, 13426—13430
[13] Yu Y. G., Kim K. S., Jin B. S.,PeptidesforInhibitionofHIVInfection, US 6489449 B2, 2002-12-03
[14] Shi W., Qi Z., Pan C., Xue N., Asim K. D., Qie J. K., Jiang S. B., Liu K. L.,Biochem.Biophys.Res.Commun., 2008, 374(4), 767—772
[15] Shi W., Cai L., Lu L., Wang C., Wang K., Xu L., Zhang S., Han H., Jiang X., Zheng B., Jiang S., Liu K.,Chem.Commun., 2012, 48, 11579—11581
[16] Huang S., Li Z., Huang P., Chang Y., Huang P.,Biochem.Biophys.Res.Commun., 2003, 307, 1029—1037
[17] Helieh S., Jeffrey O., Ebersole L.,J.CancerTher., 2010, 1(3), 105—113
[18] Di P. R., Mazzon E., Muià C.,Genovese T., Menegazzi M., Zaffini R., Suzuski H., Cuzzocrea S.,RespirRes., 2005, 6, 66—78
[19] Massimo D., Carmela F., Roberta D., Raffaella G., Claudio G., Roberta M.,AnnIstSuperSanità, 2007, 43(4), 348—361
[20] Liu S., Lu H., Zhao Q., He Y., Niu J., Debnath A. K., Wu S., Jiang S.,Biochim.Biophys.Acta, 2005, 1723, 270—281
[21] Fassina G., Buffa A., Benelli R., Varnier O. E., Noonan D. M., Albini A.,AIDS, 2002, 16, 939— 941
[22] Lee H. S., Huang P. L., Zhang D., Lee J. W., Bao J., Sun Y., Chang Y. T., Zhang J. Z., Huang P. L.,Biochem.Biophys.Res.Commun., 2007, 354, 872—878
[23] Bao J., Zhang D. W., Zhang J. Z. H., Huang P. L., Huang P. L., Huang S. L.,FEBSLetters, 2007, 581, 2737—2742
[24] Wang C., Shi W., Cai L., Lu L., Yu F., Wang Q., Jiang X., Xu X., Wang K., Xu L., Jiang S., Liu K.,J.Antimicrob.Chemo-ther., 2014, 69, 1537—1545
[25] Jayan N., Yoshio H., Junfa F., Kenneth L. K.,Bioorg.Chem., 2003, 31, 191—197
[26] Gambacorta A., Tofani D., Bernini R., Migliorini A.,J.Agric.FoodChem., 2007, 55, 3386—3391
[27] Mary K. L., Shawn B., Kelly I. G., Teresa B. B., Stephen R. P. Jr., Gene M.,Biochemistry, 1996, 35, 13697—13708
[28] Liang G. D., Wang C., Shi W. G., Wang K., Jiang X. F., Xu X. Y., Liu K. L.,Chem.J.ChineseUniversities, 2014, 35(10), 2100—2103(梁国栋, 王潮, 史卫国, 王昆, 姜喜凤, 许笑宇, 刘克良. 高等学校化学学报, 2014, 35(10), 2100—2103)
(Ed.: P, H, Y, K)
Design, Sythesis and Biological Evaluation of Polyphenol-α-helical Peptide Conjugates as Potent HIV-1 Fusion Inhibitors†
† Supported by the National Natural Science Foundation of China(Nos.81573266, 81373266).
CHENG Siqi1, LIANG Guodong2, JIANG Xifeng2, WANG Chao2*, LIU Keliang1,2*
(1.SchoolofPharmaceuticalEngineering,ShenyangPharmaceuticalUniversity,Shenyang110016,China;2.BeijingInstituteofPharmacologyandToxicology,AcademyofMilitaryMedicalSciences,Beijing100850,China)
Denovo-designed peptide sequences with anα-helical conformation can prevent fusogenic gp41 six-helical bundle(6HB) formation by specifically interacting with the Human Immunodificiency virus type 1(HIV-1) gp41 N-terminal heptad repeat(NHR) region, thus inhibiting HIV-1-cell membrane fusion. Meanwhile, polyphenols, such as hydroxytyrosol(HT) and (-)-epigallocatechin gallate(EGCG), are a major group of natural compounds with a broad spectrum of antiviral activity. In this paper, mHT were designed and obtained based on 3,4-dihydroxymethylacetic acid. Then, mHT and other bioactive polyphenols, were covalently conjugated to a certainα-helical peptide through specific linkers with different lengths and flexibilities. These conjugates interacted with the gp41 NHR region and exhibited promising fusion inhibitory activity, with IC50values in the low micromolar range. This study provides a promising strategy for the development of fusion inhibitors against viruses with class I fusion proteins.
Human immunodificiency virus type 1(HIV-1); Fusion inhibitor;α-Helical peptide; Polyphenol

2016-04-29.
日期: 2016-06-22.
国家自然科学基金(批准号: 81573266, 81373266)资助.
10.7503/cjcu20160295
O629.72
A
联系人简介: 刘克良, 男, 博士, 研究员, 博士生导师, 主要从事多肽药物、 核酸化学及药用高分子材料研究.
E-mail: keliangliu55@126.com
王 潮, 男, 博士, 助理研究员, 主要从事多肽药物研究. E-mail: chaow301@gmail.com
猜你喜欢
生物化学与生物物理进展(2022年7期)2022-07-25
生物化学与生物物理进展(2022年6期)2022-07-21
寻根(2022年2期)2022-04-17
陶瓷学报(2021年5期)2021-11-22
科学技术与工程(2021年8期)2021-04-22
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19
山东工业技术(2016年15期)2016-12-01
浙江大学学报(人文社会科学版)预印本(2016年4期)2016-02-28
浙江大学学报(人文社会科学版)预印本(2016年4期)2016-02-28
浙江大学学报(人文社会科学版)预印本(2016年4期)2016-02-28
