加速溶剂萃取—硅胶柱净化—碱性氧化铝柱分离—气相色谱三重四极杆质谱法测定土壤中的二噁英类化合物
2017-06-15 19:26邬静胡吉成马玉龙王世杰王英金军
分析化学 2017年6期
邬静+胡吉成+马玉龙+王世杰+王英+金军


摘 要 建立了加速溶剂萃取(ASE),酸性硅胶柱、复合硅胶柱及碱性氧化铝柱纯化分离,气相色谱三重四极杆质谱测定土壤中二噁英/呋喃(PCDD/Fs)、多氯联苯(PCBs)、多氯萘(PCNs)的分析方法。选用正己烷二氯甲烷(50∶50, V/V)作为ASE的提取溶剂,设定提取温度为120℃,加标回收实验表明本方法可行。用100 mL正己烷二氯甲烷(95∶5, V/V)及50 mL正己烷二氯甲烷(50∶50, V/V)依次淋洗碱性氧化铝柱,得到组分A(PCBs及PCNs)与组分B(PCDD/Fs),实现了PCDD/Fs与另外两种化合物的分离,排除了同系物间及其它杂质的干扰。使用同位素稀释气相色谱三重四极杆质谱法(GCMS/MS), 在选择反应监测(Selected reaction monitoring, SRM)模式下测定PCDD/Fs、PCBs和PCNs,3种化合物的仪器检出限(LOD)范围分别为0.04~0.25 μg/L, 0.10~0.20 μg/L和0.01~0.05 μg/L,目标物平均相对响应因子(RRF)的相对标准偏差(RSD)均小于13%。基质土加标实验中,3种化合物13C标记的同位素内标回收率的范围分别为50%~95%,51%~103%, 49%~74%。实际样品的分析结果表明,PCDD/Fs、PCBs及PCNs在土壤样品中的总含量范围分别为16.1~1148 pg/g、6.6~152.6 pg/g及10.9~99.5 pg/g,且样品测定结果与高分辨质谱测定结果相吻合。
关键词 二噁英/呋喃; 多氯联苯; 多氯萘; 前处理方法; 三重四极杆质谱
1 引 言
多氯代二苯并二噁英(Polychlorinated dibenzopdioxins, PCDDs)、多氯代二苯并呋喃(Polychlorinated dibenzofurans, PCDFs)及多氯联苯(Polychlorinated biphenyls, PCBs)是首批列入《斯德哥尔摩公约》优先控制污染物名单中的持久性有机污染物(Persistent organic pollutants, POPs)[1,2]。其中,17种2,3,7,8位全部被氯原子取代的PCDD/Fs及12种类二噁英类PCBs(Dioxinlike polychlorinated biphenyls, DLPCBs)因具有高毒性而备受关注[3]。此外,多氯萘(Polychlorinated naphthalenes, PCNs)具有与PCDD/Fs和PCBs相似的毒性效应,近年来也引起人们的关注[4,5],并于2015年列入《斯德哥尔摩公约》。研究表明,自PCBs和PCNs被禁止作为工业化学品生產后,一些工业活动已成为二噁英类化合物的重要来源。有文献报道,垃圾焚烧、金属冶炼及化工生产等都会非故意排放PCDD/Fs、PCBs、PCNs等污染物[6~8]。虽然二噁英类化合物至今仍被排放,但其在环境中处于痕量水平[9~11],导致对样品的前处理及检测受限,因此建立一种低成本、高效率的分析方法非常必要。
美国环保署(US EPA)检测PCDD/Fs、PCBs的标准方法(1613和1668)是高分辨气相色谱质谱法(HRGCHRMS)[12,13]。近年来,高分辨质谱也已被广泛应用于分析环境样品中的PCNs[10,14]。但高分辨质谱具有购置及维护费用昂贵,仪器操作复杂等缺陷,极大限制了其在环境样品分析中的应用[15]。三重四极杆质谱(GCMS/MS)购置及维护费用接近常规的低分辩质谱,并且在选择反应监测(Selected reaction monitoring, SRM)模式下,可以有效减少仪器检测过程中的背景干扰,降低检出限,适用于痕量化合物的分析[16]。已有国内学者采用此质谱对上述3种化合物分别进行了分析检测。如吴嘉嘉等[17]建立了三重四极杆质谱法测定二噁英的检测方法; 张利飞等[18]也建立了三重四极杆质谱法测定环境样品中的17种二噁英的方法,并将样品分析结果与高分辨质谱分析结果进行比对,表明三重四极杆质谱可用于环境样品中二噁英的分析检测; 王炳玲等[19]建立了超声辅助萃取三重四极杆质谱法测定室内灰尘中多氯联苯的方法; 刘芷彤等[20]建立了三重四极杆质谱法测定环境样品中20种多氯萘的方法。可见,使用三重四极杆质谱法可以有效地检测环境中二噁英类化合物。
土壤样品基质复杂,在使用三重四极杆质谱法测定时,须采用高效的提取方法,并进行严格的前处理。US EPA 1613方法[11]及1668方法[12]包含建立了对于土壤样品中PCDD/Fs和PCBs前处理的标准方法,也有研究者建立了土壤中PCNs类化合物的前处理方法[21,22]。但上述处理方法都主要对其中一类化合物进行前处理,对于土壤中PCDD/Fs、PCBs及PCNs化合物的同时前处理,并采用三重四极杆质谱法进行测定的方法,目前尚未报道。因此,本研究对土壤样品进行一次前处理,建立了三重四极杆质谱仪测定17种PCDD/Fs、12种DLPCBs及6种PCNs,共35种二噁英类化合物的方法,降低了二噁英类化合物的检测成本。
2 实验部分
2.1 仪器与试剂
Trace 1310气相色谱TSQ 8000 Evo三重四极杆质谱联用仪(美国Thermo Fisher Scientific公司); 24 LCS型冻干机(德国 Christ公司); ASE300快速溶剂萃取仪(美国Dionex公司); R3型旋转蒸发仪(瑞士Buchi公司); BF2000氮气吹干仪(北京八方世纪科技有限公司)。
正己烷、二氯甲烷(农残级,美国J.T.Baker公司); 活化硅胶(使用前于600℃马弗炉中烘烤6 h以上),酸性硅胶(79 g浓H2SO4逐滴加入到100 g活化硅胶中,振荡摇匀),碱性硅胶(1 mol/L NaOH溶液 26 g逐滴加入至50 g活化硅胶中,振荡摇匀),硝酸银硅胶(2.4 mol/L AgNO3溶液20 g逐滴加入至50 g活化硅胶中,振荡摇匀),碱性Al2O3(使用前于600℃马弗炉中烘烤6 h以上),无水Na2SO4(使用前于500℃马弗炉中烘烤6 h以上)。
PCDD/Fs标准品(1613 STOCK:2,3,7,8TCDF、2,3,7,8TCDD、1,2,3,7,8PeCDF、2,3,4,7,8PeCDF、1,2,3,7,8PeCDD、1,2,3,4,7,8HxCDF、1,2,3,6,7,8HxCDF、2,3,4,6,7,8HxCDF、1,2,3,7,8,9HxCDF、1,2,3,4,7,8HxCDD、1,2,3,6,7,8HxCDD、1,2,3,7,8,9HxCDD、1,2,3,4,6,7,8HpCDF、1,2,3,4,7,8,9HpCDF、1,2,3,4,6,7,8HpCDD、OCDF、OCDD); 13C12PCDD/Fs(DFLCSC:与1613 STOCK中各化合物相对应的13C取代的化合物),均购自Wellingon公司; PCNs标准品(ECN5497:1,2,3,4TetraCN、1,2,3,5,7PentaCN、1,2,3,4,6,7HexaCN、1,2,3,5,6,7HexaCN、1,2,3,5,6,8HexaCN、1,2,3,4,5,6,7HeptaCN、OctaCN); 13C10PCNs(ECN5102:13C101,2,3,4TetraCN、1,3,5,7TetraCN、1,2,3,5,7PentaCN、1,2,3,5,6,7HexaCN、1,2,3,4,5,6,7HeptaCN、OctaCN),PCBs标准品(World Health Organization Congener Mix:CB77, 81, 105, 114, 118, 123, 126, 156, 157, 167, 169和189); 13C12PCBs(CB77, 81, 105, 114, 118, 123, 126, 156, 157, 167, 169, 189, 180和170)均购自Cambridge Isotope Laboratories公司。
2.2 样品前处理方法
将土壤样品冷冻干燥后,称取20.0 g,加入装好滤膜的萃取池中,用加速溶剂萃取(ASE)仪进行提取。萃取完成后将溶液转入鸡心瓶中,旋转蒸发至近干后,过酸性硅胶柱(70 mL正己烷活化,90 mL正己烷洗脱)及复合硅胶柱(70 mL正己烷活化,90 mL正己烷洗脱),再利用碱性氧化铝柱进行进一步纯化分离(湿法填柱),加入样品后先用100 mL二氯甲烷正己烷(5∶95, V/V)洗脱,得组分A(PCBs及PCNs),再用50 mL二氯甲烷正己烷(50∶50, V/V)洗脱,得组分B(PCDD/Fs)。
2.3 仪器条件
色谱柱:DB5MS(60 m×0.25 mm i.d. × 0.25 μm; J&W scientific公司),采用不分流進样模式,载气为高纯He,流速1.0 mL/min。
PCDD/Fs的升温程序:初始温度160℃,保持2 min; 7.5℃/min升至220℃,保持16 min; 5℃/min升至235℃,保持7 min; 5℃/min升至330℃,保持1 min。
PCBs的升温程序:初始温度120℃,保持1 min; 30℃/min升至150℃; 2.5℃/min升至300℃,保持1 min。
PCNs的升温程序:初始温度80℃,保持2 min; 20℃/min升至180℃,保持1 min; 2.5℃/min升至280℃; 10℃/min升至290℃,保持5 min。
质谱条件:电离模式EI,离子源电压70 eV,PCDD/Fs离子源温度280℃,PCNs及PCBs离子源温度270℃,采用SRM模式,具体参数见3.2节。
3 结果与讨论
3.1 前处理方法优化
3.1.1提取方法优化 快速溶剂萃取仪能够高效快速地对固体物质中的POPs进行萃取,有文献报道了利用其对土壤或其它环境介质中PCDD/Fs、PCBs及PCNs分别进行萃取的方法,萃取温度为100℃~150℃[22~24]。为同时保证3种化合物的提取效率,本研究选用120℃作为提取温度。US EPA 1613方法[11]中采用正己烷二氯甲烷(50∶50, V/V)作为目标物的提取溶剂,本研究参考此方法,设置ASE提取溶液配比为正己烷二氯甲烷(50∶50, V/V),提取液体积为萃取池体积的60%。最终,ASE萃取仪参数设置为:正己烷二氯甲烷(50∶50, V/V)提取,系统压力1500 Psi,提取温度120℃,加热6 min,静态提取5 min, 循环2次,用萃取池体积60%量的溶液冲洗,氮气吹扫时间100 s。
为验证提取方法的可行性,用无水Na2SO4代替基质土,进行提取方法的加标回收率实验(n=3),实验结果表明,PCDD/Fs各单体的平均回收率范围为73%~90%,PCBs各单体的平均回收率范围为79%~85%,PCNs各单体的平均回收率范围为76%~89%,目标物平均回收率的相对标准偏差(RSD)均小于15%,平行性良好,提取方法高效可行。
3.1.2 净化方法优化 土壤样品基质复杂,须采用多种净化柱除杂。酸性硅胶常用于去除脂肪、还原性物质等杂质,碱性硅胶用于去除酚类及磺胺类杂质,硝酸银硅胶主要用于去除样品中的含硫杂质,碱性氧化铝常被用于去除氯代联苯醚对PCDD/Fs在检测时的干扰[12,25]。本实验参考US EPA 1613方法及1668方法对土壤样品的前处理方法,最终选用酸性硅胶柱、复合硅胶柱及碱性氧化铝纯化分离土壤样品中PCDD/Fs、PCBs及PCNs(具体柱参数如图1所示)。
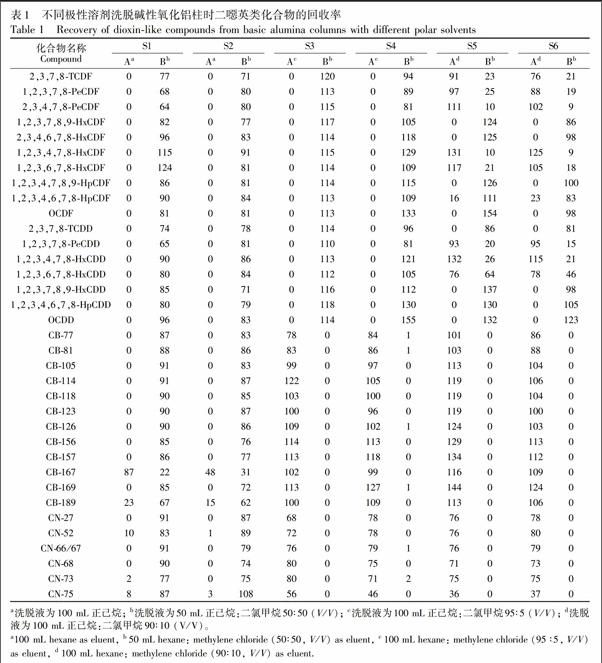
已有研究表明,即使使用高分辨质谱,也很难消除PCBs与PCDD/Fs的相互干扰[26],因此在实验中,常用合适的纯化分离柱将PCBs与PCDD/Fs分成不同组分,以便检测分析[23,25]。有文献报道使用多段硅胶柱氧化铝柱活性炭柱流程净化并分离PCDD/Fs与PCBs[23],但所用溶剂甲苯对身体不利。本研究采用碱性氧化铝柱将PCDD/Fs与其它两类化合物分离,避免其在三重四极杆质谱检测过程中的干扰。通过柱头加标实验(PCDD/Fs、PCBs和PCNs加标量均为5 ng),探讨了二噁英类化合物纯化分离的方法,样品(S1~S6)中各化合物回收率如表1所示。
首先,参照US EPA 1613方法,用100 mL正己烷洗脱碱性氧化铝柱,洗脱液作为组分A,再用50 mL二氯甲烷正己烷(50∶50, V/V)冲洗,洗脱液作为组分B,实验设置2个平行样(S1、S2),浓缩后进行GCMS/MS分析并计算各化合物平均回收率。实验结果表明,在两个平行样的组分A中,只有CB189及CB167流出,平均回收率分别为19%和68%。在组分B中3种化合物的检出率均为100%,回收率为22%~124%,说明正己烷作为洗脱溶剂,不能将PCDD/Fs与其它两种化合物分离。
增大溶剂极性,分别采用正己烷二氯甲烷(95∶5, V/V)及正己烷二氯甲烷(90∶10, V/V)洗脱碱性氧化铝柱,洗脱液作为组分A,再用正己烷二氯甲烷(50∶50, V/V)洗脱,洗脱液作为组分B,不同溶剂极性设置2个平行样(S3, S4及S5, S6),浓缩后进行GCMS/MS分析并计算回收率。使用上述两种溶液配比洗脱过程中,组分A中PCBs单体的回收率为78%~144%,PCNs单体的回收率为36%~80%,组分B中2种化合物单体回收率均小于2%。说明增大洗脱液极性,可以将PCBs与PCNs洗脱入组分A中。但在使用正己烷二氯甲烷(90∶10, V/V)洗脱过程中,2,3,7,8TCDF, 1,2,3,7,8PeCDF, 2,3,4,7,8PeCDF, 1,2,3,7,8PeCDD, 1,2,3,4,7,8HxCDF, 1,2,3,6,7,8HxCDF, 1,2,3,4,7,8HxCDD, 1,2,3,6,7,8HxCDD和1,2,3,4,6,7,8HpCDF也流入组分A中,在组分A中回收率为16%~132%,说明在第一次洗脱碱性氧化铝柱的过程中,溶剂极性过大会导致部分PCDD/Fs被提前洗脱下来。
综合以上实验结果, 建立了利用碱性氧化铝将PCBs、PCNs与PCDD/Fs类化合物纯化分离的方法,首先采用100 mL正己烷二氯甲烷(95∶5, V/V)洗脱组分A(PCBs及PCNs),再用50 mL二氯甲烷正己烷(50∶50, V/V)洗脱组分B(PCDD/Fs),有效减少了3种化合物在质谱分析过程中的相互干扰。具体前处理流程见图2。
3.2 质谱方法建立
3.2.1 质谱条件选择与优化 已有文献分别报道了PCDD/Fs[17]、PCBs[16]及PCNs[20]的三重四极杆质谱测定方法。本实验在已有文献的基础上,优化了部分质谱参数,使目标化合物的仪器响应达到最大。首先分别用3种化合物的标准溶液进行全扫描分析,获得全扫描质谱图。考察各化合物的碎片离子及相对丰度,每种单体选择两个质荷比大且相对丰度高的碎片离子作为母離子。结果表明,除五氯代多氯联苯(Pentachlorinated biphenyls, PentaCBs)、13C10CN27、CN52及CN73外,其它化合物单体的选择离子均与文献中一致。本研究中PentaCBs的定量及定性离子对分别为m/z 323.9/254.0及m/z 325.9/256.0,13C10CN27的定量及定性离子对分别为m/z 275.9/204.0及m/z 275.9/206.0,CN52的定量及定性离子对分别为m/z 301.9/229.8及m/z 299.8/227.8,CN73的定量及定性离子对分别为m/z 367.8/297.8及369.8/299.8。
3.2.2 碰撞电压优化 设定碰撞电压16, 18, 20, 25, 30, 35, 40, 45, 50, 55和60 eV序列,优化碰撞电压,使每种化合物的子离子响应最大。优化后,TeCDFs、HxCDFs、PeCDFs、HpCDFs、PCDDs、PCBs和PCNs的碰撞电压为30、30、25、25、18、25和30 eV。PCDD/Fs、PCBs及PCNs的标准品选择反应监测总离子流图如图3。
3.3 方法验证
3.3.1 平均相对响应因子(RRF)、检出限(LOD)及定量限(LOQ) 利用上述三重四极杆质谱SRM方法,将PCDD/Fs, PCBs和PCNs的标准校正溶液分别进样(其中TCDD/DFs、PeHpCDD/DFs、OCDD/DF、PCBs和PCNs的校正溶液浓度范围分别为0.2~80.0 ng/mL、1.0~400.0 ng/mL、2.0~800.0 ng/mL、 6.4~500.0 ng/mL、1.0~400.0 ng/mL),根据US EPA 1613方法[12],计算平均相对响应因子和RSD,结果如表2所示。配制系列浓度梯度接近检出限的标准溶液样品,采用已建立的质谱方法进行分析,以3倍信噪比确定各化合物单体的LOD,以10倍信噪比确定各化合物的LOQ,结果见表2。
3.3.2 基质加标实验 采集土壤,用快速溶剂萃取仪对土壤样品进行预提取,作为基质土使用。以每份15.0 g基质土,称取3份平行样,加入13C标记的PCDD/Fs、PCBs及PCNs同位素定量内标溶液,利用本方法对样品进行分析测定。3个平行样中各化合物的回收率及相对标准偏差见表3。样品中PCDD/Fs, PCBs及PCNs同位素定量内标溶液平均回收率分别为50%~95%,51%~103%及49%~74%,满足痕量分析的要求。回收率RSD范围为3%~30%,平行性较好。
3.4 实际样品分析
利用本方法对某再生铜厂与再生铝厂附近6份土壤样品及1份空白样品进行测定。空白中未检出PCDD/Fs, PCBs及PCNs。6份土壤样品中PCDD/Fs, PCBs及PCNs 的总浓度范围分别为16.1~1148 pg/g, 6.6~152.6 pg/g及10.9~99.5 pg/g。相关研究表明,PCDD/Fs及PCBs的浓度越低,三重四极杆质谱和高分辨质谱测定的结果差异性越大[22,27]。因此,本实验选择上述6个样品中浓度水平较低的样品,采用高分辨质谱测定其3种化合物的含量水平,与三重四极杆质谱测定结果进行比较。对比三重四极杆质谱与高分辨质谱测定各化合物单体的结果(图4)可知,两种方法的测定结果具有一致性(R2=0.94~0.99)。
綜上所述,快速溶剂萃取,酸性硅胶柱、复合硅胶柱及碱性氧化铝柱净化分离,同位素稀释三重四极杆质谱法可以作为同时测定土壤中PCDD/Fs、PCBs及PCNs的有效方法。
4 结 论
建立了快速溶剂萃取,酸性硅胶、复合硅胶柱及碱性氧化铝柱纯化分离, 气相色谱三重四极杆质谱同时测定土壤样品中17种PCDD/Fs、12种DLPCBs及6种PCNs的方法。ASE法可方便快捷地提取出土壤样品中的3种二噁英类化合物; 酸性硅胶柱、复合硅胶柱及碱性氧化铝柱可将提取后的样品进行净化,将PCDD/Fs与PCBs、PCNs分离,降低了三重四极杆质谱检测时3种化合物的相互干扰。本方法对土壤样品一次前处理,低分辨质谱测定PCDD/Fs、PCBs及PCNs,是一种低成本、高效率的新方法。
References
1 Berg M V D, Birnbaum L S, Denison M, Vito D M, Farland W, Feeley M, Fiedler H, Hakansson H, Hanberg A, Haws L, Rose M, Safe S, Schrenk D, Tohyama C, Tritscher A, Tuomisto J, Tysklind M, Walker N, Peterson R E. Toxicol. Sci., 2006, 93(2): 223-241
2 XU PengJun, TAO Bu, LI Nan, ZHENG Sen, ZHAO Hu, FAN Shuang, ZHOU ZhiGuang, REN Yue, QI Li, CHEN JiPing. Chinese J. Anal. Chem., 2015, 43(3): 356-365
许鹏军, 陶 晡, 李 楠, 郑 森, 赵 虎, 范 爽, 周志广, 任 玥, 齐 丽, 陈吉平. 分析化学, 2015, 43(3): 356-365
3 Kulkarni P S, Crespo J G, Afonso C A M. Environ. Int., 2008, 34(1): 139-153
4 LI YanJing, REN MingZhong, ZHANG ManWen, ZHANG SuKun. Journal of Instrumental Analysis, 2010, 29(Suppl.): 219-222
李艳静, 任明忠, 张漫雯, 张素坤. 分析测试学报, 2010, 29(增刊): 219-222
5 Kucklick J R, Helm P A. Anal. Bioanal. Chem., 2006, 386(4): 819-836
6 Nie Z Q, Liu G R, Liu W B, Zhang B, Zheng M H. Atmo. Environ., 2012, 57: 109-115
7 Li S, Liu G R, Zheng M H, Liu W B, Wang M, Xiao K, Li C L, Wang Y W. Chemosphere, 2015, 126: 73-77
8 Meng B, Ma W L, Liu L Y, Zhu N Z, Song W W, Lo C Y, Li J, Kannan K, Li Y F. Atmos. Pollut. Res., 2016, 7(2): 355-362
9 Spongberg A L, Witter J D. Rev. Biol. Trop., 2008, 56(4): 1-9
10 GUO Li, BA Te, ZHENG MingHui. Prog. Chem., 2009, 21(2/3): 377-388
郭 丽, 巴 特, 郑明辉. 化学进展, 2009, 21(2/3): 377-388
11 Zieliński M, Kamińska J, Czerska M, Ligocka D, Urbaniak M. Int. J. Occup. Med. Environ. Health., 2014, 27(6): 902-918
12 Method 1613B. Tetrathrough Octachlorinated Dioxins and Furans by Isotope Dilution HRGC/HRMS. US EPA1997
13 Method 1668A. Chlorinated Biphenyl Congeners in Water, Soil, Sediment, Biosolids, and Tissue by HRGC/HRMS. US EPA1999
14 Lega R, Megson D, Hartley C, Crozier P, Macpherson K, Kolic T, Helm P A, Myers A, Bhavsar S P, Reiner E J. J. Chromatogr. A, 2017, 1479: 169-176
15 Reiner E J. Mass. Spectrom. Rev., 2010, 29(4): 526-559
16 GarciaBermejo A, Abalos M, Saulo J, Abad E, Gonzalez M J, Gomara B. Anal. Chim. Acta, 2015, 889: 156-165
17 WU JiaJia, ZHANG Bing, DONG ShuJun, ZHENG MingHui. Chinese J. Anal. Chem., 2011, 39(9): 1297-1301
吳嘉嘉, 张 兵, 董姝君, 郑明辉. 分析化学, 2011, 39(9): 1297-1301
18 ZHANG LiFei, REN Yue, XU PengJun, DONG Liang, LIU AiMin, WU ZhongXiang, HUANG YeRu. China Environ. Sci., 2014, 34(4): 1052-1058
张利飞, 任 玥, 许鹏军, 董 亮, 刘爱民, 吴忠祥, 黄业茹. 中国环境科学, 2014, 34(4): 1052-1058
19 WANG BingLing, ZHANG XiaoLing, ZHANG Qi, LU XiaoMei, CUI Yuan, ZHANG ZhengDong. Chinese Journal of Chromatography, 2014, 32(1): 74-80
王炳玲, 张晓玲, 张 琦, 陆晓梅, 崔 媛, 张正东. 色谱, 2014, 32(1): 74-80
20 LIU ZhiTong, ZHANG Bing, WANG WenWen, LIU GuoRui, GAO LiRong, ZHENG MingHui. Chinese Journal of Chromatography, 2013, 31(9): 878-884
刘芷彤, 张 兵, 王雯雯, 刘国瑞, 高丽荣, 郑明辉. 色谱, 2013, 31(9): 878-884
21 Nadal M, Schuhmacher M, Domingo J L. Chemosphere, 2007, 66(2): 267-276
22 ZHANG LiFei, ZHANG XiuLan, ZHANG Hui, LI LingLing, ZHANG LinLi, DONG Liang. Chinese J. Anal. Chem., 2014, 42(2): 258-266张利飞, 张秀蓝, 张 辉, 李玲玲, 张琳利, 董 亮. 分析化学, 2014, 42(2): 258-266
23 LI Wei, CHEN ZuoSheng, LI ChangQing, HUANG Ping, LIU GengYun, ZHOU Zheng, WANG GuanYu. Res. Environ. Sci., 2004, 17(2): 61-64
李 伟, 陈左生, 李常青, 黄 萍, 刘耕耘, 周 正, 王关玉. 环境科学研究, 2004, 17(2): 61-64
24 SHAO Yang, YANG GuoSheng, HAN Shen, MA LingLing, LUO Min, LIU WeiHua, XU DianDou. Chinese J. Anal. Chem., 2016, 44(5): 698-706
邵 阳, 杨国胜, 韩 深, 马玲玲, 罗 敏, 刘韦华, 徐殿斗. 分析化学, 2016, 44(5): 698-706
25 NING HuiPing, ZHANG ManWen, REN MingZhong, WANG Zheng, ZHANG SuKun. Environ. Chem., 2012, 31(7): 1110-1111
宁慧平, 张漫雯, 任明忠, 王 拯, 张素坤. 环境化学, 2012, 31(7): 1110-1111
26 Asplund L, Grafstrm A K, Haglund P, Jansson B, Jrnberg U, Mace D, Strandell M, Wit C D. Chemosphere, 1990, 20(1012): 1481-1488
27 Dam T G, Pussente I C, Scholl G, Eppe G, Schaechtele A, Leeuwen V S. J. Chromatogr. A, 2016, 1477: 76-90
Abstract A method for determination of PCDD/Fs, PCBs and PCNs in soil sample was developed by using accelerated solvent extraction (ASE)silica gel column cleanupbasic alumina column separation coupled with GCMS/MS. The sample was extracted by ASE with Hexanemethylene chloride (HexDCM, 50∶50, V/V) at 120℃. The basic alumina column was used to separate PCDD/Fs, PCBs and PCNs. The extracts were eluted with HexDCM (95∶5, V/V) to obtain PCBs and PCNs, followed by HexDCM (50∶50, V/V) to obtain PCDD/Fs. The limits of detection (LOD) were in the range of 0.04-0.25 μg/L, 0.10-0.20 μg/L and 0.01-0.05 μg/L for PCDD/Fs, PCBs, PCNs, respectively. The relative standard deviations (RSDs) of average relative response factors (RRF) were below 13%. The recoveries of 13Clabeled internal standards of the three classes of analytes were 50%-95%, 51%-103% and 49%-74%, respectively. Concentrations of ∑PCDD/Fs, ∑PCBs and ∑PCNs in soil samples were 16.1-1148 pg/g, 6.6-152.6 pg/g and 10.9-99.5 pg/g, respectively. The results were consistent with that of high resolution mass spectrometer.
Keywords Polychlorinated dibenzopdioxins and furans; Polychlorinated biphenyls; Polychlorinated naphthalenes; Pretreatment methods; Triple quadrupole mass spectrometry
(Received 10 January 2017; accepted 5 April 2017)
This work was supported by the National Natural Science Foundation of China (No. 21407185), the Fundamental Research Funds for the Central Universities (Nos. 2017MDYL31, 2015MDTD23C), the Institution of Higher Education Innovation Talent Recruitment Program (No. B08044), and the National College Students' Innovative Training Program (No. GCCX2016110042)