
PAMAM树状大分子负载和释放阿霉素的耗散粒子动力学模拟
2017-10-13 15:31苏运祥全学波闵文凤乔来聪李理波周健
化工学报 2017年5期
苏运祥,全学波,闵文凤,乔来聪,李理波,周健
PAMAM树状大分子负载和释放阿霉素的耗散粒子动力学模拟
苏运祥,全学波,闵文凤,乔来聪,李理波,周健
(华南理工大学化学与化工学院,广东省绿色化学产品技术重点实验室,广东广州510640)
采用耗散粒子动力学模拟方法研究了药物输送载体聚酰胺-胺(PAMAM)树状大分子对抗癌药物阿霉素(DOX)的负载和释放行为。构建了PAMAM树状大分子的粗粒化模型,该模型能准确地重现树状大分子的构象性质。考察了PAMAM树状大分子代数(G)对DOX负载以及pH环境对DOX释放的影响。模拟结果表明,PAMAM树状大分子主要通过疏水作用将DOX包封于内部空腔,G6和G7 PAMAM树状大分子的负载能力较强,因为其孔隙率较高,内部有更多的疏水空腔。在低pH环境下,PAMAM树状大分子结构发生变化,DOX分子能快速地从其中释放,主要原因是PAMAM的伯胺、叔胺和DOX伯胺发生质子化,质子化基团间的静电排斥作用使得PAMAM树状大分子发生溶胀,导致其内部空腔暴露,促进了DOX的释放。本工作可以为基于树状大分子的药物输送体系的设计和优化提供参考。
药物输送;树状大分子;包封;模型;介尺度;分子模拟
引 言
目前癌症已经成为威胁人类健康的主要杀手之一,癌症治疗的方法主要有手术切除、放疗和化疗。化疗是属于全身治疗的方法,其治疗剂量和频率由于恶心、脱发、骨髓抑制、免疫力下降、心脏毒性等副作用而受到限制[1]。此外,大多数化疗药物水溶性差,难以被吸收利用。因此,开发新型的药物输运载体来提高抗癌药物的溶解度和降低其毒副作用显得十分重要,要解决这些问题首先需要理解载体和药物分子间的相互作用机制。
树状大分子是一类新型的人工合成的具有超支化结构的高分子聚合物[2-5],它具有高度对称的球形结构,其结构可分为3部分:内核、内部重复亚单元、末端基构成的壳。其中,连接中心核的重复亚单元向外发散开,组成一系列同心圈层,也就是树状大分子的代数(G),树状大分子的内核被称为第0代(G0),重复亚单元组成的同心圈层依次为第1,2,…,代(Gn)。由于它是逐步合成的,所以其分子大小、形状和官能团可被精确控制。此外,树状大分子良好的水溶性和生物相容性使其成为理想的药物输送载体[6-8],近年来它已经被用于化疗药物的负载研究。Yellepeddi等[9]研究了PAMAM/顺氯氨铂复合物的毒性和毒性机理,顺氯氨铂在PAMAM树状大分子中的负载量接近11%(mass),结果表明PAMAM树状大分子可作为顺氯氨铂的潜在输送载体治疗卵巢癌。Ooya等[10]采用聚丙三醇(PGDs)树状大分子作为紫杉醇(PTX)的增溶剂,与单纯的药物相比,通过PGDs树状大分子包封,PTX的水溶性增强了400倍。Markatou等[11]将二甲氧基姜黄素分别与G3.5和G4代PAMAM树状大分子混合以提高二甲氧基姜黄素的水溶性,它们对二甲氧基姜黄素的最大包封率可达到90%和95%。Kojima等[12]将分子量为550和2000的聚乙二醇(PEG)单甲醚分别接枝到G3和G4代PAMAM树状大分子表面,其对阿霉素(DOX)和甲氨蝶呤(MTX)的包封能力随着PAMAM代数和PEG接枝链长增长而增强。Bhadra等[13]采用表面PEG化的G4代PAMAM树状大分子负载5-氟尿嘧啶,发现表面PEG化不但可以增强PAMAM树状大分子的药物负载能力,还可以降低药物释放的速率和溶血毒性。
虽然实验上对树状大分子负载药物分子进行了大量研究,但是对其包封和释放的机理认识还不足。这方面可以通过计算机分子模拟增强理解,当前的模拟研究主要是采用分子动力学(MD)研究药物分子与PAMAM树状大分子的相互作用[14-17]。如Shi等[15]采用MD模拟了以羧基为端基的PAMAM树状大分子与2-甲氧基雌二醇的复合物结构;pH为5时,PAMAM的羧基与其内部的叔氨基间的强静电作用使复合物的结构很紧密,导致2-甲氧基雌二醇无法释放,失去生物活性。虽然MD能够研究复合物的结构及它们的形成驱动力,但是难以得到药物复合和释放的整个动态过程,因为研究这一过程需要更大更长时空尺度的模拟方法。耗散粒子动力学(dissipation particle dynamics, DPD)就是其中一种,自1992年由Hoogerbrugge等[18]提出以来,已被广泛地用于聚合物体系自组装和药物负载行为研究[19-21]。本文将采用DPD模拟方法研究PAMAM树状大分子和DOX在水溶液中的自组装,考察PAMAM代数对DOX负载及pH环境对DOX释放的影响,从介尺度揭示PAMAM树状大分子载体对药物分子的负载和释放机理。
1 模拟方法
1.1 DPD基本原理
DPD模拟采用粗粒化模型,体系由一系列质量和体积相同的软相互作用粒子组成。1个DPD珠子对应若干个小分子或高分子的几个链段。所有珠子的运动遵守牛顿运动方程[22]
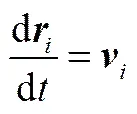
(2)
其中,、、m、分别表示珠子的位置矢量、速度、质量和总的作用力。为了简化计算,在DPD中物理量均采用约化单位(reduced unit)。所有珠子的质量设定为1 DPD单位。珠子受到的总作用力包括保守力(C)、耗散力(D)、随机力(R)、键作用力(S)、静电力(E)5种[23]

珠子之间的相互作用力在超过截断半径(c)时消失,DPD中c作为长度的约化单位,取c=1。前3种力的大小和方向由珠子位置和速度决定,其计算公式如下
(4)
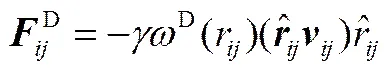
(6)
式中,a代表珠子和之间的排斥参数;=-,r=||,,=-;为噪声振幅;为耗散力系数;是平均值为0的随机涨落变量;D(r)和R(r)为权重函数。两个权重函数的关系及选取满足式(7),而噪声振幅和耗散力系数存在式(8)的关系
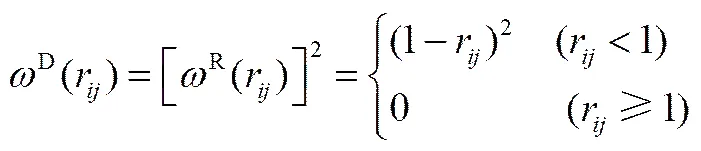
2=2kB(8)
式中,B为Boltzmann常数;为热力学温度。在DPD模拟中,B为能量约化单位,取B=1。耗散力系数取值4.5,从而=3。同一分子中相连接的两个成键珠子之间存在弹簧力,表示为
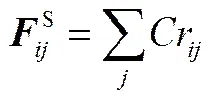
式中,为弹簧力常数,取值=4。
静电力E的求解采用Ewald sum方法[24],表示为
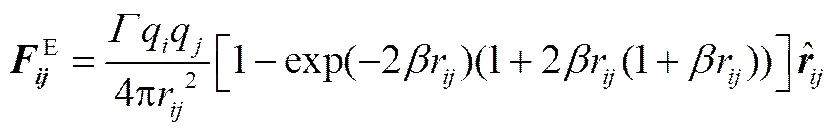
式中,=2/(B0rc),为电子电量,0为真空介电常数,r为室温时水的介电常数;为珠子电荷数;=c/,为电荷衰减长度。
1.2 模型和相互作用参数
本文模拟的体系由PAMAM树状大分子、药物DOX和水组成,该体系的粗粒化模型如图1所示。PAMAM树状大分子由3种珠子构成,分别标记为P1、P2和P3;其中,P1珠子代表N—(CH2)3片段,P2珠子代表CH2—C(O)—NH—CH2片段,P3珠子代表CH2—NH2片段,图1(a)所示的是G1 代的PAMAM,更高代的PAMAM分子以相同的方式构建。模型药物DOX也由3种珠子构成,分别标记为D1、D2、D3,1个水分子作为1个W珠子。PAMAM树状大分子的伯胺基(P3珠子)和叔氨基(P1珠子),以及DOX分子的伯胺基(D3珠子)在不同的pH环境下有不同的解离状态。在生理pH(约7.4)下,所有的P3珠子质子化,带1个单位正电荷;在低pH(约5)时,所有的P1、P3和D3珠子质子化,都带1个单位正电荷[25-26]。
在DPD模拟中,不同珠子间的保守力排斥参数a取决于相对应原子的相互作用,与Flory-Huggins ()参数满足如下线性关系[27]
a=a+ 3.27(11)
式中,a为相同珠子间的排斥参数,可由Groot等[28]给出的公式计算
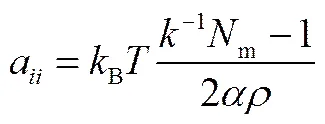
式中,-1为常温常压下水的量纲1压缩因子,其值约为15.98;m表示1个DPD水珠子所代表的水分子数;常数=0.101±0.001;为体系的珠子数密度。当=1,m=1,=3时,计算得到a=25。不同珠子间的参数可通过计算混合能求出
(13)
式中,是配位数;E是组分和组分的混合能;为理想气体常数;为热力学温度。本文采用Materials Studio 4.4软件中的blends模块在298 K的温度下,选择COMPASS力场计算得到参数。每一对组分的混合能都是通过Monte Carlo方法产生的2000000个不同构型进行平均得到的。最后通过式(11)计算得到不同珠子间的排斥参数a,其值如表1所示。由于P1、P3和D3珠子在不同pH时发生质子化而带上正电,故加入一定量的反号离子C以保持体系的电中性。
表1 不同珠子之间的排斥参数aij
Table 1 Repulsive parameters aij between different beads

表1 不同珠子之间的排斥参数aij
aijD1D2D3P1P2P3WC D125.00 D268.8825.00 D383.5332.9525.00 P172.4131.5424.6725.00 P276.3711.9234.9441.5425.00 P3114.4031.4727.3824.5840.8925.00 W150.8044.4439.7930.1335.2025.0425.00 C25.0025.0025.0025.0025.0025.0025.0025.00
本文进行DPD模拟所采用的是DL_MESO 2.5模拟软件包[29]。长程静电力采用Ewald sum方法处理,为避免带电珠子重叠在一起,采用slater-type电荷密度分布,smearing系数=0.929,静电截断半径Cele=3,实空间收敛常数取值0.97,倒易空间向量max=(5,5,5),介电耦合常数=13.87。模拟盒子大小为30×30×303C,由于珠子数密度选为3,体系的DPD珠子总数为81000个,盒子的、、3个坐标方向上均采用周期性边界条件。模拟积分时间步长为0.05,药物负载和释放的模拟步数分别为100000和30000步。珠子质子化后与其他珠子间的排斥参数不变,只是其带电量改变。
2 结果与讨论
2.1 PAMAM树状大分子的构象性质
树状大分子的构象性质可由回转半径(g)定性描述,对于一个含有个粗粒化珠子的PAMAM树状大分子,其g可由式(14)求出
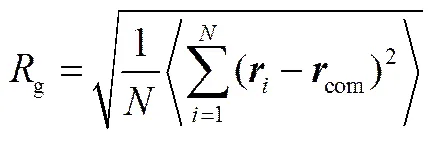
式中,为珠子的位置矢量;com为PAMAM分子质心位置矢量。本文模拟体系中每个珠子的平均体积约为0.03 nm3,由于珠子的数密度为3,因此3C= 0.09,从而可计算出C=0.45 nm。为了验证PAMAM树状大分子的粗粒化模型是否合理,本文模拟计算了代数为G3到G7的PAMAM树状大分子在溶液中的g,结果列于表2。为了比较,表2也列出了Rathgeber等[30]采用小角度X射线散射(SAXS)获得的实验值和Maiti等[31]采用全原子MD得到的模拟值。从表2可观察到本DPD模拟计算得到的g值与SAXS实验值和MD模拟值基本一致,这表明PAMAM的CG模型是准确可靠的。因此,在接下来的模拟中,将采用该CG模型研究G3~G7代的PAMAM树状大分子对DOX的负载和释放过程。
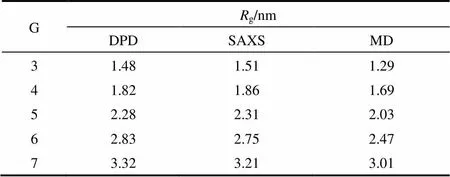
表2 不同代数的PAMAM树状大分子的回转半径以及SAXS实验值和MD模拟值
2.2 PAMAM树状大分子对DOX负载过程和药物分布
PAMAM树状大分子可作为单分子胶束,将小分子药物溶解包封于其内部空腔。本模拟采用DPD研究了G5代PAMAM树状大分子负载药物时对DOX的包封动态过程,如图2所示。模拟体系中放入1个G5代的PAMAM树状大分子,DOX和水的体积分数分别为0.4%和99%。为了清楚地显示分子聚集形貌,水珠子W未显示。
由图2 可以看出,模拟开始时DOX分子随机地分布在水中[图2(a)]。随着模拟时间的增加,一部分DOX分子逐渐地扩散进入PAMAM分子的内部空腔;同时,另一部分DOX由于自身的疏水性互相聚集在一起[图2(b)~(f)]。此后,继续增加模拟时间,PAMAM载药胶束和DOX分子的聚集形态均未发生较大变化[图2(g)~(h)]。因此,100000步的时间足以使模拟达到平衡状态。进一步研究了体系达到平衡后,PAMAM载药胶束中DOX药物的分布和DOX与PAMAM树状大分子各个珠子间的径向分布函数。胶束中PAMAM的P1、P2和P3珠子和DOX的密度分布曲线如图3所示。
从图3中可见,距离PAMAM载药胶束中心较近时,DOX的珠子密度出现峰值,而此时PAMAM的P3珠子未出现,当P3珠子的密度逐渐升高时,DOX的密度迅速下降,说明DOX主要分布在由PAMAM树状大分子的P1和P2珠子构成的内部空腔中。
径向分布函数(RDF)提供了胶束中不同珠子间的相对位置信息,珠子间的RDF出现峰值时的距离近则表明它们亲和性好,所以可以用RDF来分析不同组分的相容性。PAMAM树状大分子的径向分布函数如图4所示。从图4(a)可以看出,D1珠子与P1和P2珠子的径向分布函数的峰值较高,而与P3珠子的则较低,且出现峰值时D1珠子与P3珠子的距离较D1与P1和P2珠子的距离远,说明D1珠子与P1和P2珠子的相容性较好。D2和D3珠子与PAMAM各个珠子间的径向分布函数曲线与D1珠子的相似[图4(b)、(c)],说明P1和P2珠子与DOX分子的各珠子相容性较好,而P3珠子与DOX珠子的相容性则较差,这和表1中PAMAM树状大分子与DOX分子各珠子间的排斥参数a相一致。
此外由表1 还以看出,P1和P2珠子与水珠子W间的排斥参数较大(30.13和35.20),而P3珠子的则较小(25.04),说明P1和P2珠子较疏水,P3珠子较亲水,同时DOX分子的3种珠子也较疏水(与W的排斥参数分别为150.80、44.44和39.79),这充分证明DOX分子主要是靠疏水作用聚集在PAMAM树状大分子的内部疏水空腔中。
2.3 PAMAM树状大分子代数对DOX负载的影响
不同代数的PAMAM树状大分子具有不同的分子构象,其内部空腔的大小和数量也不同,所以PAMMA树状大分子的代数会影响其对DOX分子的包封能力。G3~G7代的PAMAM树状大分子对DOX的包封平衡构象如图5所示。各模拟体系中均放入1个PAMAM树状大分子,药物分子的体积分数固定为0.4%,其余均为水分子。从图5中可以看出,随着PAMAM树状大分子代数的增加,更多的DOX被包封进PAMAM树状大分子中。
包封率可以用来衡量PAMAM树状大分子的包封能力,其定义为被负载的DOX分子占体系总的DOX分子的百分比。经计算,G3~G7 PAMAM树状大分子对DOX的包封率分别为21.1%、56.6%、65.7%、92.5%和100%。
由于DOX主要包封于PAMAM树状大分子的内部空腔中,内部空腔的体积直接影响PAMAM的载药量,为了定量地表征PAMAM树状大分子的空腔体积的含量,本文计算了G3~G7代的PAMAM树状大分子的孔隙率[32],如图6 所示。PAMAM树状大分子的孔隙率定义为其内部空腔体积占分子体积的百分比。为计算PAMAM树状大分子的内部空腔体积,假设PAMAM是一个球体模型,其任一珠子的半径为c,用一个球形溶剂探针在PAMAM表面滚动,球形探针中心所形成的轨迹就是PAMMA的溶剂可及表面积(SASA),SASA表面包围的体积则为溶剂可及体积(SAV),其计算公式如下
SAV=(4p/3)(+p)3(15)
式中,为PAMAM球体模型的半径;p为探针的半径。在voss volume voxelator软件包[33]中采用不同半径的探针(p的值为1c到8c),可以计算得到一系列的SAV值。由于较大的探针分子无法探测到PAMAM的空腔,而较小的探针则可以探测到空腔,所以将用较大半径的探针得到的PAMAM分子的SAV值做一线性拟合,得到的拟合值与计算值之差即为PAMAM的空腔体积。
从图6可以看出,G3代PAMAM树状大分子的孔隙率较低,所以其对DOX的包封能力较差,包封率仅有21.1%,而G5 PAMAM树状大分子的孔隙率比G4的稍低,所以其对DOX的包封率略高于G4代PAMAM 树状大分子(包封率分别为65.7%和56.6%),G6和G7 代PAMAM的树状大分子的孔隙率较高,包封能力较强(包封率分别为92.5%和100%),所以负载药物时,采用G6或G7代PAMAM 树状大分子负载DOX较好。因此,下面将主要研究G6代PAMAM 树状大分子对药物的释放过程。
2.4 pH对药物释放的影响
PAMAM 树状大分子的伯胺基和叔胺基,以及DOX分子的伯胺基在不同的pH环境下发生质子化,从而改变分子的结构。本DPD模拟研究了生理pH和低pH时,G6代PAMAM树状大分子释放DOX药物的动态过程。模拟体系中放入一个G6 代的PAMAM树状大分子,药物的体积分数为0.4%,其余为水分子和反号离子。模拟结果分别如图7和图8所示。从图7可以看出,在生理pH时,没有DOX分子从G6代PAMAM树状大分子中释出,这是因为PAMAM树状大分子发生溶胀,分子由致密的核结构变为致密的壳结构,而致密的壳结构抑制了DOX从PAMAM树状大分子的空腔中释放。这有利于载药胶束在体内循环时保持其稳定性。
低pH时,DOX从G6代PAMAM树状大分子中释放的动态过程如图8所示。模拟以G6 代PAMAM树状大分子在负载药物平衡后的构型作为释放过程的初始构型[图8(a)]。模拟时间为2000 步时,G6代PAMAM树状大分子的结构不再发生大的变化,此时有少量的DOX分子释出[图8(b)]。进一步增加模拟时间,不断有DOX从PAMAM树状大分子中释放[图8(c)~(e)],模拟时间为30000步时,DOX药物全部释放[图8(f)]。药物的释放主要是由PAMAM 树状大分子的P1和P3珠子以及DOX的D3珠子质子化造成的。一方面P1和P3珠子带正电后,带电珠子间相互产生排斥,使PAMAM树状大分子结构发生较大变化。图9所示的是在不同pH环境下,G6代PAMAM 树状大分子的回转半径随模拟时间的变化。从图中可以观察到,在低pH时G6代PAMAM树状大分子的回转半径要比其在生理pH时的大26.7%,说明低pH时,PAMAM树状大分子发生了更大的溶胀,这使得其内部疏水空腔直接暴露于外部溶液环境。另一方面,PAMAM树状大分子P1、P2珠子与DOX的D3珠子间的静电排斥作用进一步促进了DOX分子从PAMAM树状大分子中释放出来。
3 结 论
本文采用耗散粒子动力学模拟的方法研究了PAMAM树状大分子在水溶液中对DOX的包封和释放行为。首先构建了G3~G7代PAMAM树状大分子的粗粒化模型,通过与SAXS实验和其他MD模拟对比,发现该模型能很好地重现PAMAM树状大分子的构象性质。模拟结果表明DOX药物主要分布在由P1和P2珠子构成内部空腔中,对珠子间的径向分布函数和排斥参数分析表明PAMAM主要是通过疏水作用将DOX包封于其内部疏水空腔中。模拟发现G6和G7代PAMAM树状大分子对DOX的包封率分别为92.5%和100%,通过对G3~G7代PAMAM树状大分子孔隙率的分析,发现G6和G7代PAMAM树状大分子对DOX的包封能力较强,是因为其孔隙率较高,相同体积情况下,内部有更多的空腔可用于负载疏水药物。模拟揭示了DOX在两种pH环境下的释放动态过程。在生理pH条件下,没有DOX分子从G6代PAMAM树状大分子中释出,但是PAMAM树状大分子发生溶胀,分子由致密的核结构变为致密的壳结构,抑制药物释放。在低pH时,DOX分子能快速地从PAMAM树状大分子中释出,主要是因为PAMAM 树状大分子的伯胺(P3珠子)和叔氨基(P1珠子)以及DOX分子的伯胺基(D3珠子)发生了质子化,一方面使得PAMAM 树状大分子进一步溶胀,导致其内部疏水空腔暴露于外部溶液环境,另一方面质子化基团间的静电排斥进一步促进了DOX分子的释放。本文模拟结果可为基于树状大分子的药物输送体系的设计和优化提供有价值的参考。
符 号 说 明

aij——珠子i、j之间的DPD排斥参数,kJ·mol-1·nm-2 C——同一分子链中两个成键珠子之间的弹簧力常数,kJ·mol-1·nm-2 Eij——珠子i、j之间的混合能,kcal·mol-1 e——元电荷,=1.602176565 ×10-19 C FCij——珠子i、j之间的保守力,N FDij——珠子i、j之间的耗散力,N FEij——珠子i、j之间的静电力,N FRij——珠子i、j之间的随机力,N FSij——相连珠子i、j之间的键作用力,N fi——珠子i所受到的总作用力,N k-1——常温常压下水的量纲1压缩因子 kB——Boltzmann常数,J·K-1 kmax——倒易空间矢量 mi——珠子i的质量,kg N——树状大分子所含珠子数 Nm——1个水珠子所代表的水分子数 q——珠子电荷量,C R——理想气体常数,R=8.314 J·mol-1·K-1 Rg——回转半径,nm rCele——珠子静电截断半径,nm rc——珠子截断半径,nm rcom——树状大分子质心矢量,nm ri——珠子i的位置矢量,nm T——热力学温度,K vi——珠子i的速度,m·s-1 G——介电耦合常数 a——实空间收敛常数 b——slater-type电荷密度分布涂布系数 cij——珠子i、j之间的Flory-Huggins参数 er——室温时水的介电常数,F·m-1 e0——真空介电常数,F·m-1 g——耗散力系数 qij——均值为0的随机涨落变量 r——珠子数密度 s——噪声振幅 wD(rij)——耗散力权重函数 wR(rij)——随机力权重函数 下角标 i,j——珠子
References
[1] FOX M E, SZOKA F C, FRÉCHET J M J. Soluble polymer carriers for the treatment of cancer: the importance of molecular architecture[J]. Acc. Chem. Res., 2009, 42(8): 1141-1151.
[2] TOMALIA D A, BAKER H, DEWALD J,. A new class of polymers: starburst-dendritic macromolecules[J]. Polym. J., 1985, 17(1): 117-132.
[3] WU P, FELDMAN A K, NUGENT A K,. Efficiency and fidelity in a click-chemistry route to triazole dendrimers by the copper(Ⅰ)-catalyzed ligation of azides and alkynes[J]. Angew. Chem. Int. Ed., 2004, 43(30): 3928-3932.
[4] CARNAHAN M A, GRINSTAFF M W. Synthesis of generational polyester dendrimers derived from glycerol and succinic or adipic acid[J]. Macromolecules, 2006, 39(2): 609-616.
[5] ZHONG T P, AI P F, ZHOU J. Structures and properties of PAMAM dendrimer: a multi-scale simulation study[J]. Fluid Phase Equilib., 2011, 302(1/2): 43-47.
[6] GILLIES E R, FRÉCHET J M J. Dendrimers and dendritic polymers in drug delivery[J]. Drug Discovery Today, 2005, 10(1): 35-43.
[7] GUPTA U, AGASHE H B, ASTHANA A,. Dendrimers: novel polymeric nanoarchitectures for solubility enhancement[J]. Biomacromolecules, 2006, 7(3): 649-658.
[8] CHENG Y Y, WANG J R, RAO T L,. Pharmaceutical applications of dendrimers: promising nanocarriers for drug delivery[J]. Frontiers in Bioscience-Landmark, 2008, 13: 1447-1471.
[9] YELLEPEDDI V K, VANGARA K K, PALAKURTHI S. Poly(amido)amine (PAMAM) dendrimer-cisplatin complexes for chemotherapy of cisplatin-resistant ovarian cancer cells[J]. J. Nanopart. Res., 2013, 15(9): 1-15.
[10] OOYA T, LEE J, PARK K. Hydrotropic dendrimers of generations 4 and 5: synthesis, characterization, and hydrotropic solubilization of paclitaxel[J]. Bioconjugate Chem., 2004, 15(6): 1221-1229.
[11] MARKATOU E, GIONIS V, CHRYSSIKOS G D,. Molecular interactions between dimethoxycurcumin and Pamam dendrimer carriers[J]. International Journal of Pharmaceutics, 2007, 339(1/2): 231-236.
[12] KOJIMA C, KONO K, MARUYAMA K,. Synthesis of polyamidoamine dendrimers having poly(ethylene glycol) grafts and their ability to encapsulate anticancer drugs[J]. Bioconjugate Chem., 2000, 11(6): 910-917.
[13] BHADRA D, BHADRA S, JAIN S,. A PEGylated dendritic nanoparticulate carrier of fluorouracil[J]. International Journal of Pharmaceutics, 2003, 257(1/2): 111-124.
[14] TANIS I, KARATASOS K. Association of a weakly acidic anti-inflammatory drug (ibuprofen) with a poly(amidoamine) dendrimer as studied by molecular dynamics simulations[J]. J. Phys. Chem. B, 2009, 113(31): 10984-10993.
[15] SHI X, LEE I, CHEN X,. Influence of dendrimer surface charge on the bioactivity of 2-methoxyestradiol complexed with dendrimers[J]. Soft Matter, 2010, 6(11): 2539-2545.
[16] ABDERREZAK A, BOURASSA P, MANDEVILLE J-S,. Dendrimers bind antioxidant polyphenols and-platin drug[J]. PLoS ONE, 2012, 7(3): e33102.
[17] MAINGI V, KUMAR M V S, MAITI P K. PAMAM dendrimer-drug interactions: effect of pH on the binding and release pattern[J]. J. Phys. Chem. B, 2012, 116(14): 4370-4376.
[18] HOOGERBRUGGE P J, KOELMAN J M V A. Simulating microscopic hydrodynamic phenomena with dissipative particle dynamics[J]. Europhys. Lett., 1992, 19(3): 155-160.
[19] 郭泓雨, 崔洁铭, 孙德林, 等. 温敏性两亲嵌段共聚物相行为的耗散粒子动力学模拟[J]. 化工学报, 2012, 63(11): 3707-3715. GUO H Y, CUI J M, SUN D L,. Dissipative particle dynamics simulation on phase behavior of thermo-responsive amphiphilic copolymer PCL-PNIPAM-PCL[J]. CIESC Journal, 2012, 63(11): 3707-3715.
[20] 孙德林, 周健. 耗散粒子动力学模拟Nafion膜和PVA/Nafion共混膜的介观结构[J]. 物理化学学报, 2012, 28(4): 909-916. SUN D L, ZHOU J. Dissipative particle dynamics simulations on mesoscopic structures of Nafion and PVA/Nafion blend membranes [J]. Acta Physica-Chimica Sinica, 2012, 28(14): 909-916.
[21] 刘红艳, 郭泓雨, 周健. PLGA-PEG共聚物负载多西紫杉醇的药物输运体系的计算机模拟[J]. 化学学报, 2012, 70(23): 2445-2450. LIU H Y, GUO H Y, ZHOU J. Computer simulations on the anticancer drug delivery system of docetaxel and PLGA-PEG copolymer[J]. Acta Chimica Sinica, 2012, 70(23): 2445-2450.
[22] GROOT R D, WARREN P B. Dissipative particle dynamics: bridging the gap between atomistic and mesoscopic simulation[J]. J. Chem. Phys., 1997, 107(11): 4423-4435.
[23] GROOT R D. Electrostatic interactions in dissipative particle dynamics-simulation of polyelectrolytes and anionic surfactants[J]. J. Chem. Phys., 2003, 118(24): 11265-11277.
[24] GONZ LEZ-MELCHOR M, MAYORAL E, VEL ZQUEZ M E,. Electrostatic interactions in dissipative particle dynamics using the Ewald sums[J]. J. Chem. Phys., 2006, 125(22): 224107.
[25] LIN S T, MAITI P K, GODDARD W A. Dynamics and thermodynamics of water in PAMAM dendrimers at subnanosecond time scales[J]. J. Phys. Chem. B, 2005, 109(18): 8663-8672.
[26] WEN X F, LAN J L, CAI Z Q,. Dissipative particle dynamics simulation on drug loading/release in polyester-PEG dendrimer[J]. J. Nanopart. Res., 2014, 16(5): 2403.
[27] GROOT R D, MADDEN T J. Dynamic simulation of diblock copolymer microphase separation[J]. J. Chem. Phys., 1998, 108(20): 8713-8724.
[28] GROOT R D, RABONE K L. Mesoscopic simulation of cell membrane damage, morphology change and rupture by nonionic surfactants[J]. Biophys. J., 2001, 81(2): 725-736.
[29] SEATON M A, ANDERSON R L, METZ S,. DL_MESO: highly scalable mesoscale simulations[J]. Molecular Simulation, 2013, 39(10): 796-821.
[30] RATHGEBER S, MONKENBUSCH M, KREITSCHMANN M,. Dynamics of star-burst dendrimers in solution in relation to their structural properties[J]. J. Chem. Phys., 2002, 117(8): 4047-4062.
[31] MAITI P K, MESSINA R. Counterion distribution and ζ-potential in PAMAM dendrimer[J]. Macromolecules, 2008, 41(13): 5002-5006.
[32] MAITI P K, CAGIN T, WANG G F,. Structure of PAMAM dendrimers: generations 1 through 11[J]. Macromolecules, 2004, 37(16): 6236-6254.
[33] VOSS N R, GERSTEIN M, STEITZ T A,. The geometry of the ribosomal polypeptide exit tunnel[J]. J. Mol. Biol., 2006, 360(4): 893-906.
Dissipative particle dynamics simulations on loading and release of doxorubicin by PAMAM dendrimers
SU Yunxiang, QUAN Xuebo, MIN Wenfeng, QIAO Laicong, LI Libo, ZHOU Jian
(School of Chemistry and Chemical Engineering, Guangdong Provincial Key Laboratory for Green Chemical Product Technology, South China University of Technology, Guangzhou 510640, Guangdong, China)
Dissipative particle dynamics (DPD) simulations were employed to study the loading and release behaviors of anticancer drug doxorubicin (DOX) by drug delivery carrier polyamidoamine (PAMAM) dendrimers. A coarse-grained (CG) model for PAMAM dendrimers was first constructed, which reproduced the conformational properties of PAMAM dendrimers accurately. The effects of PAMAM dendrimer generation (G) on DOX loading and the environment pH on DOX release were investigated. Simulation results showed that PAMAM dendrimers mainly encapsulated DOX into their interior cavities through hydrophobic interaction. The encapsulation capacity of G6 and G7 PAMAM dendrimers were much better than PAMAM of lower generations, because there were more hydrophobic cavities inside G6 or G7 dendrimers for their high porosity. At low pH, PAMAM dendrimers underwent conformational changes, thus DOX molecule escaped from dendrimers quickly. Such phenomena are mainly caused by the protonation of primary amines and tertiary amines in PAMAM dendrimers and primary amines in DOX. The electrostatic repulsion between these charged groups will lead PAMAM dendrimers swelling immensely and the inner cavities being exposed, which promotes the release of DOX molecules. This work could provide useful guidance for the design and optimization of dendrimer-based drug delivery systems.
drug delivery; dendrimer; encapsulation; model; mesoscale; molecular simulation
10.11949/j.issn.0438-1157.20161736
O 641.3
A
0438—1157(2017)05—1757—10
周健。
苏运祥(1990—),男,硕士研究生。
国家自然科学基金项目(91334202, 21376089);国家重点基础研究发展计划项目(2013CB733500);广东省自然科学基金项目(2014A030312007);中央高校基本科研业务费项目(SCUT-2015ZP033)。
2016-12-12收到初稿,2017-02-15收到修改稿。
2016-12-12.
Prof. ZHOU Jian, jianzhou@scut.edu.cn
supported by the National Natural Science Foundation of China(91334202, 21376089), the National Basic Research Program of China(2013CB733500), the Natural Science Foundation of Guangdong Province(2014A030312007) and the Fundamental Research Founds for the Central Universities (SCUT-2015ZP033).
猜你喜欢
高等学校化学学报(2022年4期)2022-06-10
油气·石油与天然气科学(2021年9期)2021-10-10
中学生物学(2019年1期)2019-01-13
西南石油大学学报(自然科学版)(2018年6期)2018-12-26
现代园艺(2017年22期)2018-01-19
花卉(2017年22期)2017-02-25
西安文理学院学报(自然科学版)(2016年4期)2016-12-19
浙江大学学报(理学版)(2016年6期)2016-12-15
中国烟草学报(2016年3期)2016-11-23
中学生数理化·中考版(2015年10期)2015-09-10
