
钠离子电池合金类负极材料的研究进展
2017-11-04 08:15卢海燕曹余良
中国材料进展 2017年10期
刘 创,卢海燕,曹余良
(武汉大学化学与分子科学学院,湖北 武汉 430072)
特约专栏
2017-08-09
国家重点基础研究发展计划资助项目(2016YFB0901500)和国家自然科学基金资助项目(21333007,21673165)
刘 创,男,1993年生,硕士研究生
曹余良,男,1974年生,教授,博士生导师,E-mail:ylcao@whu.edu.cn
10.7502/j.issn.1674-3962.2017.10.05
钠离子电池合金类负极材料的研究进展
刘 创,卢海燕,曹余良
(武汉大学化学与分子科学学院,湖北 武汉 430072)
钠离子电池由于具有钠资源丰富、价格低廉等特点,逐渐成为大规模储能领域的研究热点。而负极材料作为钠离子电池的重要组成部分,对电池性能有着直接的影响。在目前所研究的负极材料中,合金类负极材料由于具有较高的理论比容量而受到了人们的广泛关注。但是在反应过程中,合金类材料面临着严重的体积膨胀问题,循环稳定性差,这制约了其发展。研究者通过包覆、纳米化、与其他金属复合等方法大大改善了合金类材料的循环性能。简介了合金类储钠负极材料Sn、Sb、P、Ge、Bi、Pb和Si的反应机理及研究进展,探讨了合金类负极材料所面临的问题和解决办法,并对合金类负极材料的发展方向进行了展望。
钠离子电池;负极;合金;机理;锑;锡;磷
1 前 言
能源问题和环境问题严重威胁到了人类的生存和发展,因此人类必须开发新的可再生清洁能源来代替不可再生的化石能源,这样才能支撑人类文明的可持续发展。而各种可再生能源如太阳能、风能、水能和潮汐能等的使用需要相应的储能技术来保证能量的连续稳定输出,因此发展先进的储能技术迫在眉睫。目前储能技术种类繁多,包括物理储能、化学储能、相变储能和电磁储能等[1]。在这些储能技术当中,化学储能系统具有高效、便捷、安全、清洁等优点,在新能源技术发展中占据着重要的地位[2]。在各种化学储能中,锂离子电池能量密度高,技术十分成熟。但是随着新能源汽车的兴起,锂资源的需求量大大增加,有限的锂资源使得锂离子电池无法应用于大规模储能领域[3]。而与Li同主族的Na元素,有着与Li十分相似的物理化学性质,并且在地壳中储量丰富(2.74%),价格低廉,这使得钠离子电池十分适合大规模的储能领域。
目前钠离子电池所面临的主要问题是要找到合适的电极材料[4],常见的正极材料有过渡金属氧化物类、普鲁士蓝类化合物和聚阴离子类化合物等[5, 6],而常见的负极材料有碳基材料、合金类材料、钛基材料和有机化合物材料等[3, 5]。其中,碳基材料具有来源广泛、制备简单、环境友好和价格低廉等优点,是一种十分有应用前景的储钠负极材料。然而碳基材料比容量较低、首周不可逆容量大、倍率性能差,这制约了其发展。而合金类负极拥有高的理论比容量和良好的倍率性能,是一类非常有潜力的储钠负极材料。
本文简述了各类合金类负极材料合金化反应机理,并针对目前的研究进展,分析了所存在的问题和发展趋势。
2 合金类负极
目前,研究过的合金类材料主要有Sn、Sb、P、Ge、Bi、Pb和Si等。这类材料与Na发生合金化反应的一般方程式为:
M+xNa+ +xe↔NaxM
(1)
由于是具有多电子的合金化反应,其理论比容量一般比较高,然而高的合金化容量也造成循环过程中体积膨胀严重 (其理论比容量和体积膨胀率如图1所示),这会导致活性物质粉化脱离电接触,进而引起容量快速衰减。解决上述问题的办法主要有以下几类:①纳米化。纳米化能够减小颗粒表面的应力,同时减小钠离子的扩散距离;②包覆。将导电材料包覆在合金材料表面,提高材料的电导率,缓解其体积膨胀;③与其他金属复合,引入缓冲介质,抑制体积膨胀,增加导电性。从图1中可以发现,Bi、Ge和Pb的理论比容量较低;Si在常温电化学环境中几乎不能与钠形成合金,仅表现出吸附行为;而Sb、Sn和P三类合金材料具有较高的理论比容量(首周充放电曲线见图2)。从图3可以看出,当以Na3V2(PO4)3作正极时,Sb与C能量密度相当,但是Sb具有更高的嵌入电势(相对于金属钠电位),更加安全。而Sn和P则拥有更高的理论比容量和能量密度,因此,Sb、Sn和P是三类十分有应用前景的合金类负极材料。本文将简介上述7种材料的研究进展,并着重介绍Sb、Sn和P 3种合金类负极材料的反应机理及研究进展。
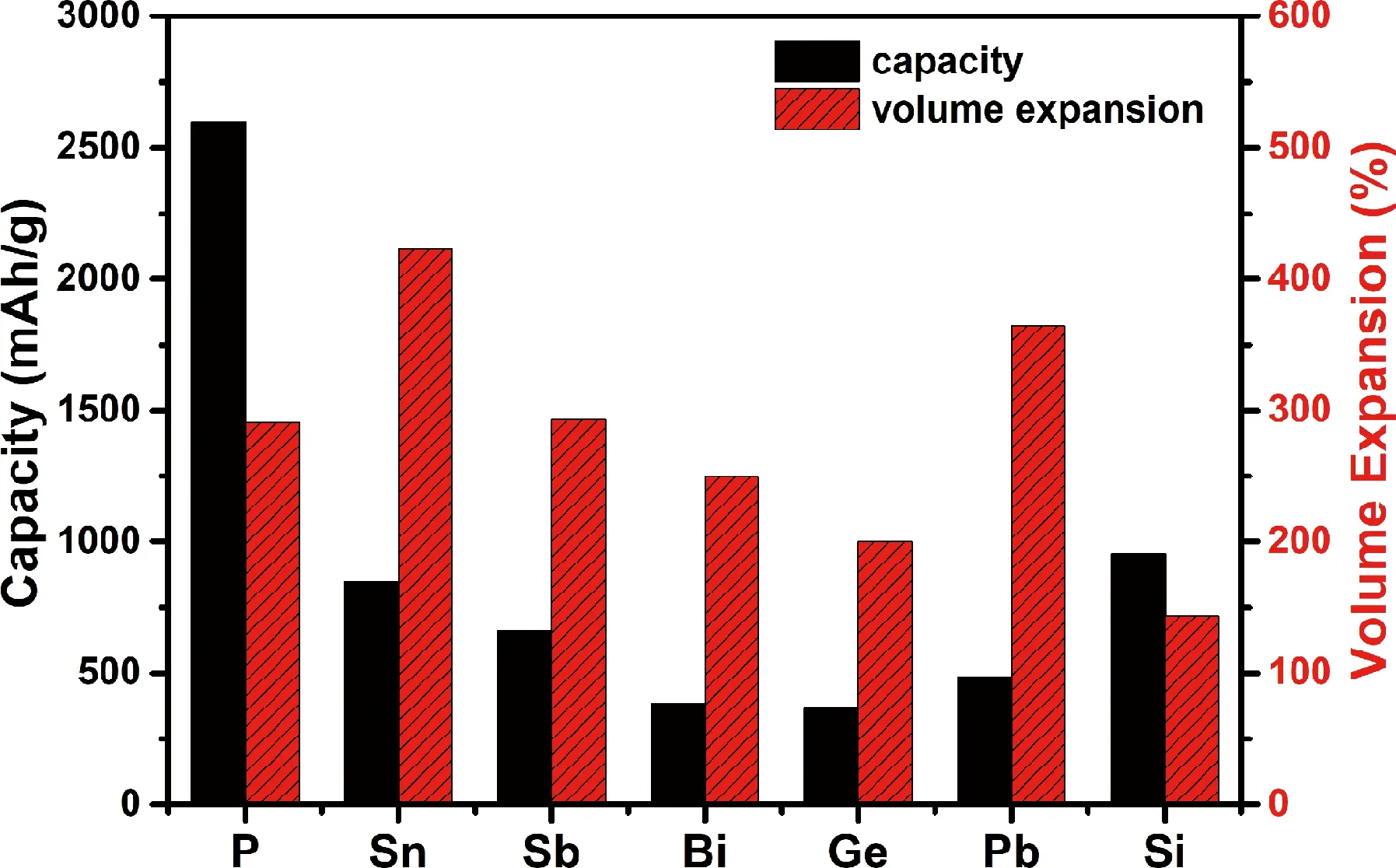
图1 合金类负极材料的理论比容量与体积膨胀率对比Fig.1 Theoretical capacities and volume expansion rates of alloy anode materials
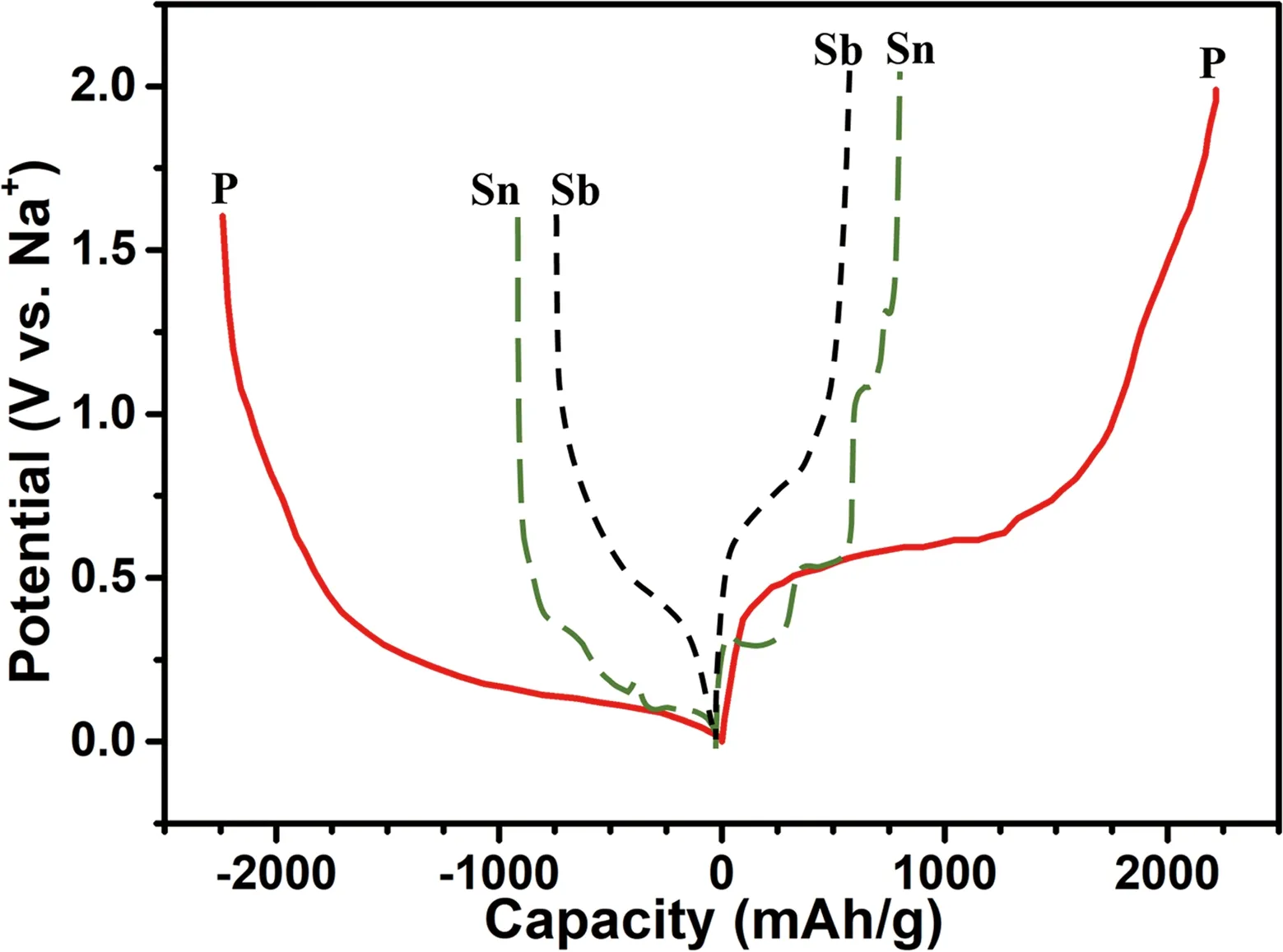
图2 Sb、Sn和P 3种合金类负极材料的首周充放电曲线Fig.2 Profile of the first charge and discharge curve of Sb, Sn and P
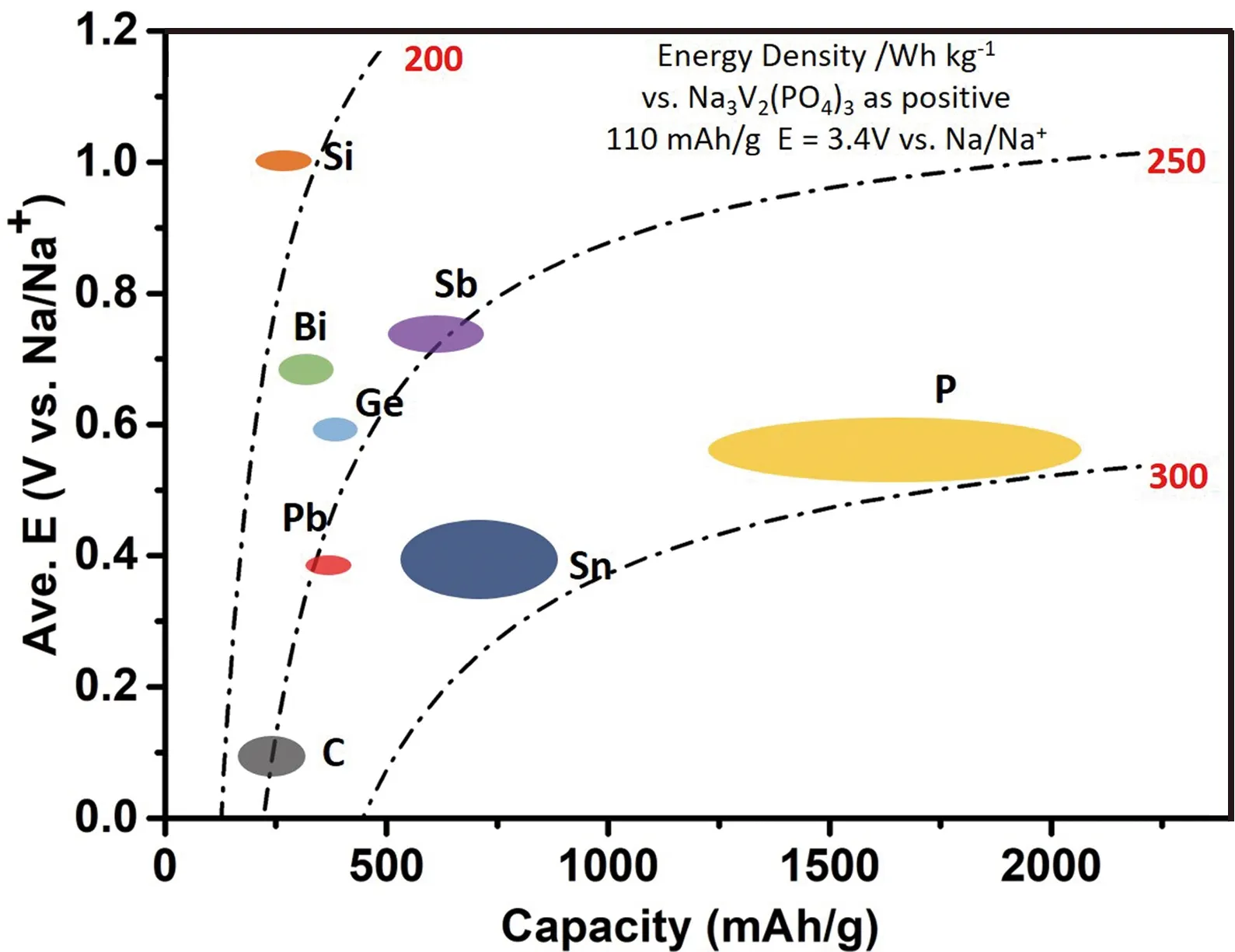
图3 合金类负极材料与碳负极材料的能量密度Fig.3 Energy density of alloy anode materials and carbon-based materials
2.1 Sb基材料
Sb是一种非常有潜力的储钠负极材料,受到了很多研究者的关注。Sb可以和Na形成NaSb和Na3Sb两种相[7](如图4a所示),理论比容量为660 mAh/g,完全嵌钠后体积膨胀率为293%。金属Sb具有褶皱的层状结构,原子堆积的空间利用率为39%,这有利于Na在层间的迁移[8],同时Sb有较好的导电性,电导率为2.5×104S/cm,有利于电子的转移,这些性能都有利于Sb的合金化反应。
对于Sb基材料体积膨胀严重,容量衰减快的问题,研究者主要采用了3类办法来提高其循环性能:① Sb-C复合物。将Sb与导电性较好的C复合,C还能提供结构支撑,缓解体积膨胀问题;② Sb-M复合物。将Sb与其他金属复合,缓解体积效应;③ 通过特殊的结构设计(微纳结构),同时保证其高的比容量和优异的循环稳定性。
2.1.1 Sb基材料的机理
Qian等[9]认为在Sb基材料充放电过程中会形成NaSb和Na3Sb,对应CV曲线上的两对氧化还原峰。但是 Darwiche等[10]通过原位XRD分析并没有发现NaSb相,而是发现了亚稳态的立方Na3Sb相的存在。Darwiche等人认为Sb基材料合金化反应过程如下:
第一次放电:c-Sb → a-NaxSb → Na3Sbhex/c-Na3Sbcub→ c-Na3Sbhex
(2)
第一次充电:c-Na3Sbhex→ a-Sb
(3)
随后放电过程:a-Sb → a-NaxSb → Na3Sbhex/ c-Na3Sbcub→ c-Na3Sbhex
(4)
随后充电过程:c-Na3Sbhex→ a-Sb
(5)
Darwiche认为Sb-Na体系在循环过程中出现的无定型相起到了一个缓解材料内部应力的作用,有利于提高材料的循环性能。而Baggetto等[11]认为Sb-Na体系的良好循环性能是由于形成了更薄更稳定的SEI膜,因此循环性能更好。同时Baggetto将Sb-Na体系较低的比容量归咎于Sb-Na体系较差的动力学性能和无定型相的存在。通过进一步研究,Baggetto等[12]认为没有形成NaSb相是由于Na原子迁移速率太慢,无法形成结构复杂的NaSb相。
Allan等[13]利用对分布函数分析(PDF)和固体核磁共振(ssNMR)等技术从另一个角度阐述了他们对Sb-Na反应机理的见解。通过对比分析,他们分离出了两个无定型中间相,分别为a-Na1.7Sb和a-Na3-xSb(0.4 2.1.2 Sb-C复合物 常见的Sb-C复合方法包括以下几种:①将Sb分散于一维碳纤维碳纳米棒之中[14, 15];②将Sb分散于C微球内部[16-18];③将Sb分散于3维碳网络[19, 20];④将Sb与高导电性的石墨烯或碳纳米管复合[21, 22];⑤制备核壳结构的Sb-C复合物[23, 24]。 Qian等[9]利用高能球磨的方法得到了Sb/C复合物,该材料中的Sb的可逆比容量高达610 mAh/g,首周库伦效率为85%,循环100周后容量保持率为94%。Wu等[25]利用静电纺丝制备的Sb-C纳米纤维在40 mA/g的电流密度下首周充电比容量为631 mAh/g,在200 mA/g的电流密度下循环400周后容量保持率为90%。Luo等[26]将Sb纳米颗粒嵌入三维碳网络中得到SbNPs@3D-C材料,该材料首周库伦效率为79.1%,可逆比容量为456 mAh/g,在100 mA/g的电流密度下循环500周后容量保持率为94.3%。 2.1.3 Sb-M复合物 Sb和其他金属的复合物是研究的较早的一类材料,根据复合的金属是否有储钠能力分为电化学惰性金属和电化学活性金属。 电化学惰性金属有Mo[27]、Al[28]、Fe[29-31]、Ni[29, 32]、Cu[33, 34]和Zn[35, 36]等。Baggetto等[33]利用磁控溅射的方法制备了Cu2Sb薄膜,该材料首周充电比容量为250 mAh/g,循环10周后容量保持率为70%。Darwiche等[37]通过球磨方法制备的FeSb2材料首周充电比容量为540 mAh/g,在300 mA/g的电流密度下循环130周后仍保持440 mAh/g的可逆比容量。 具有嵌钠电化学活性的金属有Sn、Bi和Ge等,目前研究过的有SnSb[38-41]、Sn-Ge-Sb[42]、Bi-Sb[43]、Sn-Sb-P[44]和Sn-Bi-Sb[45]等。Xiao等[41]首次利用简单的球磨方法制备了SnSb/C复合材料,该材料在100 mA/g的电流密度下循环50周后仍然有80%的容量保持率。Xie等[45]制备了Sn-Bi-Sb材料,其中Sn、Bi和Sb含量分别为10%、10%和80%。该材料在200 mA/g的电流密度下首周充电比容量为592 mAh/g,循环100周后可逆比容量为621 mAh/g。 2.1.4 Sb金属微纳结构 对于纯Sb材料,可以通过微纳结构设计来缓解体积变化。目前已制备了单分散的纳米Sb颗粒[46]、Sb空心纳米球[47]、Sb多孔空心微球[48]、柏树叶状的Sb[49]、Sb纳米棒阵列[50]、多孔Sb[51]和金属Sb纳米薄片[52]等。 Liu等[51]利用Al30Sb70制备的多孔Sb呈珊瑚状,该材料首周充电比容量高达630 mAh/g,在100 mA/g的电流密度下循环200周仍然有573.8 mAh/g的可逆比容量,这种多孔结构能够减小钠离子的传输距离,同时还有足够的机械强度使得材料在循环过程中不会粉化脱落。由于Sb具有褶皱的层状结构,层间作用力较弱,因而可以像石墨烯一样作为一种二维层状材料。受此启发,Gu等[52]利用液相剥离法制备出了只有4 nm厚的金属Sb纳米薄片,并将其作为一种新型的储钠材料。在0.1 mA/cm2的电流密度下,该材料具有1226 mAh/cm3的可逆比容量。在4 mA/cm2的电流密度还能有112 mAh/cm3的可逆比容量,并能稳定循环100周。二维Sb材料的发现为Sb基材料的研究提供了新的思路。 目前大部分Sb基材料循环稳定性还需要进一步提高,由于加入了其他惰性物质,电极材料的比容量只能到达400 mAh/g以上,纯Sb金属材料有比较好的循环稳定性以及高达500 mAh/g以上的比容量,但是制备过程较为复杂,成本较高。 金属Sn也是一种重要的储钠负极材料,可以和Na形成多种合金相[53](如图4b所示)。Sn完全钠化以后形成Na15Sn4相,体积膨胀率为423%,理论比容量为847 mAh/g,远远高于储钠碳基材料的比容量。同时Sn安全无毒,储量丰富,应用前景十分广阔。Sn基材料面临的问题与Sb基材料类似,因而解决方法也类似。 2.2.1 Sn基材料的机理 Chevrier等[54]根据密度泛函理论计算了Sn的嵌钠电势,计算结果显示有4个平台,作者认为分别对应NaSn5、NaSn、Na9Sn4和Na15Sn44种晶相。Komaba等[55]通过XRD证明了Na15Sn4晶相的存在。而Ellis等[53]也观察到了4个平台,他们通过原位XRD、DFT计算和电量分析认为4个平台分别对应如下反应: 平台1: Na + Sn → NaSn3 (6) 平台2: Na + NaSn3→ a-NaSn (7) 平台3: 5Na + 4(a-NaSn) → Na9Sn4 (8) 平台4: 6Na + Na9Sn4→ Na15Sn4 (9) 同时Ellis还证明了无定型相NaSn的存在,并认为这是由于在材料内部原子流动性较差引起的。Wang等[56]通过原位TEM直接观察了纳米Sn在充放电过程中的变化。他们观察到Sn嵌钠过程分两步,首先通过一个两相反应生成无定型NaxSn合金(x~0.5),无定型合金再经过一个单相反应进一步嵌钠形成晶态Na15Sn4。 Baggetto等[57]研究了不同厚度的Sn薄膜的反应机理,并发现截止电压、电极厚度、电流密度和循环周数不同,反应机理也不同。同时作者还发现了新的Na0.6Sn相、无定型Na1.2Sn相和六方Na5Sn2相的存在。通过进一步研究,Baggetto等[58]又发现了新相Na7Sn3,但是并没有发现Na9Sn4相的存在,作者认为这是因为形成Na9Sn4的反应动力学过慢。而在Du等[59]的研究中,XRD结果显示了Na9Sn4相的存在。最近,Stratford等[60]利用XRD、固体核磁共振(ssNMR)、穆斯堡尔谱和对分布函数分析等手段也对Sn-Na体系的反应机理进行了更加细致的研究,并分离出了大量的中间态产物,结果十分复杂。 对材料反应机理的研究能够为材料的设计提供理论支持,但是由于Sn-Na合金相较多,中间相为无定型相,难以确定其组成和结构,反应机理又会受到材料形貌、颗粒大小、微观结构和电流密度等众多因素的影响,要探究其反应机理十分困难。研究者目前还没有得出较为一致的结论,这还需要进一步研究。 2.2.2 Sn-C复合物 Sn-C复合的方法可分为制备一维纳米纤维或纳米棒[61-63]、制备C微球[64-66]和与石墨烯复合[67, 68]等。 Zhu等[61]将Sn沉积在木质纤维基质的表面,形成一层Sn薄膜,木质纤维的多孔结构有利于钠离子扩散,在嵌钠过程中,柔性的木质纤维能够形成有褶皱的表面,缓解材料内部的机械应力。这种材料在84 mA/g的电流密度下首周充电比容量达到339 mAh/g,400周后还有240 mAh/g的可逆比容量。由于沉积上去的Sn较少,因而其可逆比容量不高。Liu等[62]利用静电纺丝的方法,合成了纳米Sn和氮掺杂多孔碳纤维的复合物。该材料理论容量为645 mAh/g,在200 mA/g的电流密度下首周充电容量高达631.2 mAh/g,在2000 mA/g的大电流下循环1300周后还有483 mAh/g的可逆比容量,容量保持率90%。在10000 mA/g的电流密度下仍然有450 mAh/g的可逆比容量,显示出十分优异的循环性能和倍率性能。Mao等[63]利用静电纺丝和原子层沉积技术制备了TiO2-Sn@CNFs纳米纤维。在Sn@CNFs纳米纤维表面沉积上一层TiO2后,TiO2能够限制纳米纤维的膨胀,提供结构支撑,提高库伦效率。该材料在100 mA/g的电流密度下首周充电比容量高达610 mAh/g,在循环400周后依然有413 mAh/g的可逆比容量,显示出了较高的可逆比容量和较好的循环稳定性,但是即使是在包覆了TiO2层后,其首周库伦效率依然只有58.3%。 2.2.3 Sn-M复合物 研究者也对Sn与其他金属形成的合金的电化学性能进行了研究。已经研究过的合金有Sn-Co[69, 70]、Sn-Cu[71-73]、Sn-P[74-80]、Sn-Ni[81]、Sn-Ge[82]、Sn-Fe[83-85]、Sn-Mn[86]和Sn-Sb[38-41]等。其中研究的较多的是Sn-P和Sn-Sb复合物。 Lin等[71]利用硼氢化钠在表面活性剂存在的情况下同时还原Sn2+和Cu2+,得到了100 nm左右的Sn0.9Cu0.1纳米颗粒。该材料在169 mA/g的电流密度下循环100周后仍有420 mAh/g的可逆比容量,容量几乎没有衰减。Cu和Sn形成Cu6Sn5合金均匀地分布在Sn纳米颗粒中,一方面Cu可以提高材料的导电性,另外一方面作为惰性金属,Cu可以缓解材料嵌钠过程中体积膨胀的问题,使得材料的循环稳定性大大增强。Qian等[76]通过球磨法制备了Sn4P3/C纳米颗粒,该材料在50 mA/g的电流密度下首周充电比容量达到850 mAh/g,循环150周后容量保持率为86%。均匀分布的Sn充当了导电网络,使得P活化,而P嵌钠以后的产物Na3P又作为缓冲介质缓解Sn体积膨胀的问题,Sn和P的协同效应使得材料的循环性能大大提高。Liu等[77]合成了复杂的蛋黄-壳结构的Sn4P3@C纳米球,这种材料在100 mA/g的电流密度下首周充电比容量为790 mAh/g,在1500 mA/g(1.5 C)的电流密度下可逆比容量为505 mAh/g,循环400周后可逆比容量为360 mAh/g。但是该材料的合成过程复杂,蛋黄-壳结构具有较大的比表面积,导致其首周库伦效率较低。 2.2.4 Sn金属微纳结构 Nam等[87]利用电沉积的方法在水溶液中制备出了高纵横比的Sn纳米纤维。作者团队在水溶液中加入表面活性剂,这种表面活性剂能吸附在(200)晶面上,使得Sn定向生长形成一维的Sn纳米纤维。通过这种方法制备的Sn纳米纤维沿着相同的方向排列,呈现出一种多孔的结构。在0.001~0.65 V电压范围内,该材料在0.1 C的电流密度下循环100周后仍可输出776.26 mAh/g的可逆比容量,容量保持率为95.09%。首先,纳米纤维结构具有优异的机械稳定性;其次,多孔结构能够缓解材料嵌钠过程中的体积膨胀问题。Kim等[88]以PVDF为粘结剂制备了多孔结构的Sn电极。在0.001~1 V电压范围内,该材料在0.5 C的电流密度下首周充电比容量为674 mAh/g,首周库伦效率为63%,循环500周后仍然有519 mAh/g的可逆比容量。该材料制备方法简单,比容量高,循环性能稳定良好。但是由于该材料孔隙率过高(86%),远高于正常制备的电极材料的孔隙率(37%),因此其体积比容量较低。 Sn具有高达847 mAh/g的理论比容量,因此Sn基材料比容量容易达到500 mAh/g以上,且在大电流情况下能稳定循环超过1000周,是一种很有应用前景的储钠负极材料。 P能与Na形成Na3P合金,理论容量为2596 mAh/g,体积膨胀率为291%[89]。P有多种同素异形体,常见的有白磷、红磷和黑磷。白磷活性较高,有毒,易燃,不合适作为电极材料。红磷价格便宜、储量丰富、性质稳定,是储钠负极的理想选择。红磷主要面临的问题有3点:①电导率低(~10-14S/cm);②钠离子在红磷中迁移速度慢;③红磷嵌钠过程面临严重的体积膨胀问题。黑磷具有与石墨类似的二维层状结构,电导率高(~300 S/m)[90],离子在黑磷层间扩散快,也是一种很好的储钠材料。同时还可以剥离黑磷形成与石墨烯类似的磷烯,但是黑磷制备较为复杂。由于P的理论容量高达2596 mAh/g,因而受到了越来越多研究者的关注,从2013年起,研究者就开始研究将P作为储钠材料[91-93]。 2.3.1 P基材料的机理 由于黑磷具有层状结构,因而其首先发生嵌入反应,再发生合金化反应,理论计算和实验都证实了这一点[90, 94, 95]。黑磷X轴的通道大小为0.308 nm,足够容纳钠离子的嵌入。当钠嵌入黑磷层间后,通过XRD分析发现其层间距增大,这证明了Na的嵌入反应。由于Na的嵌入反应并没有对黑磷的结构造成重大影响,因而这部分容量可逆性较高,嵌入反应发生的电压平台在0.54~1.5 V之间。在0.54 V以下发生的是合金化反应,P-P键网络被打破,形成单独的P原子或者哑铃状的P2,形成无定型的中间相,直到最后生成Na3P相。实验发现,黑磷体积膨胀主要是在Y轴和Z轴方向上,X轴并没有发生明显的变化。据此Sun等认为,通过减小其层数将有利于缓解其体积膨胀,提高其电化学性能[90]。 2.3.2 红磷 与Sn和Sb相比,红磷除了面临更大的体积膨胀问题外,还有一个问题就是红磷电导率太低,因而大多数研究者主要是将红磷与碳复合(P-C)或者与其他金属复合形成合金(P-M)以增强其循环稳定性。目前人们研究过的合金有Zn-Ge-P[96]、Ge-P[97]、Fe-P[98, 99]、Co-P[100, 101]以及Sn-P[74-80]等。P-C复合物主要将P与石墨烯[102-104]、碳纳米管[92, 105]或者其他碳基质[91, 93, 106, 107]复合,增强材料的导电性。 Qian等[93]首次利用高能球磨法合成了无定型P/C复合物,该材料首周充电比容量高达1764 mAh/g,并且能稳定循环140周,首周库伦效率为87%。蒸发-冷凝法[108]是一种常见的制备P-C复合物的方法,Liu等[109]通过该方法将红磷均匀地分布在还原氧化石墨烯上得到P@RGO材料,该材料在1593.9 mA/g的电流密度下首周充电比容量为1211 mAh/g,循环300周后可逆比容量为914 mAh/g,在31.8 A/g的电流密度下可逆比容量仍然达到510.6 mAh/g。之前对Sn和Sb的研究中,通过微纳结构设计的纯金属材料表现出了优异的电化学性能,但是在P基材料中还很少有相关报道。最近,Zhou等[110]在甲苯溶液中通过用NaN3还原PCl5得到了P的多孔空心纳米球。该材料在520 mAh/g的电流密度下首周充电比容量为2274.5 mAh/g,在1 C的电流密度下循环600周可逆比容量为969.8 mAh/g。 2.3.3 磷烯 磷烯是只有一层或数层的黑磷,具有褶皱的层状结构,是一种新型的二维材料。常见的制备方法有机械剥离、液相剥离和化学气相沉积等[111]。Sun等[90]首次将磷烯作为储钠负极材料,三明治结构的磷烯和石墨烯复合材料在50 mA/g的电流密度下首周充电比容量高达2440 mAh/g,循环100周后容量保持率为83%。作者认为减少黑磷层数形成磷烯后,其层间距增大,有利于钠离子在层间的扩散,而石墨烯为电子的传输提供了快速通道,这使得三明治结构的磷烯-石墨烯复合材料拥有良好的电化学性能。最近,Huang等[112]利用电化学阳离子嵌入的方法制备了磷烯并将其作为储钠负极材料,该制备方法简单快速,通过调节电压可以控制磷烯的层数。通过该方法制得的磷烯在100 mA/g的电流密度下充电比容量为1968 mAh/g,50周后容量保持率为60.5%。目前磷烯在钠离子电池中的应用还不多,需要进一步探索,同时,磷烯的应用也为发展新型高性能材料提供了新思路。 P的比容量远高于Sb和Sn基材料,在保持较高的比容量的同时能稳定循环超过1000周[107, 113],然而P具有一定的毒性和可燃性,同时,其还原产物Na3P水解后会产生易燃且有毒的PH3气体,这限制了P在钠离子电池中的应用[114]。 Pb是最早研究的储钠合金负极材料,理论比容量为485 mAh/g,完全嵌钠以后体积膨胀率为365%[115]。根据Jow等[116]的研究,Pb在脱嵌钠的过程中充放电曲线有4个平台,分别对应的是NaPb3、NaPb、Na5Pb2和Na15Pb4相的形成,与Pb-Na相图相一致(如图4c所示)。Ellis等[115]利用原位XRD对Pb的充放电行为进行研究。他们通过离子溅射方法制备的Pb薄膜首周放电比容量达到484 mAh/g,非常接近Pb的理论容量。XRD结果显示第3个平台对应的是Na9Pb4相,而不是之前的研究所认为的Na5Pb2相[55, 117]。Pb在钠化过程中的反应历程为: Pb→NaPb3→NaPb→Na9Pb4→Na15Pb4 (10) 由于Pb是重金属元素,同时相对于Sb、Sn和P等材料,其理论比容量较低,这限制了其进一步发展。 图4 Sb-Na相图[7] (a), Sn-Na相图[53] (b), Pb-Na相图[115] (c), Bi-Na相图[115] (d)Fig.4 Sb-Na phase diagram[7] (a), Sn-Na phase diagram[53] (b), Pb-Na phase diagram[115] (c), Bi-Na phase diagram[115] (d) Si在锂离子电池中有很高的比容量,表现出优异的电化学性能,并有望在将来实现商业化应用。根据Na-Si相图,有4种Na-Si相被报道,分别是NaSi、NaSi2、Na4Si23和NaSi94[118],完全钠化以后形成的NaSi有高达954 mAh/g的嵌钠比容量,体积膨胀率为144%[117],因此研究者也把目光投向了储量丰富、环境友好和价格低廉的Si基材料。Komaba[55]和Ellis[115]等人首先对钠离子电池Si材料进行了研究,他们发现虽然Si有很高的理论比容量,但是实际上Si并不具有嵌钠能力。Malyi等[119]通过计算发现Na在Si中迁移的势垒高达1.14 eV,而通过掺杂能有效降低其迁移势垒。此外,根据Chevrier等[54]的计算,Si的嵌钠电势小于0.1 V。过低的嵌入电位会导致Na在电极表面析出,造成容量的衰减[120]。Jung等[121]通过理论计算发现无定形硅可以和钠形成Na0.76Si合金,比容量为725 mAh/g。Xu等[122]制备了部分无定型的Si材料,在20 mA/g的电流密度下循环100周后具有248 mAh/g的可逆比容量。同时Lim等[123]也通过机械扩散的方法制备了无定型Si-Sn材料,该材料中无定型Si具有230 mAh/g的可逆比容量。虽然无定型Si有一定储钠能力,但是其实际比容量远远低于其理论比容量,因此还需要从合金化机理入手,寻找Si基材料与Na电化学合金化的途径,以提升其实际比容量。 金属Bi也具有储钠能力,室温下能稳定存在的是NaBi和Na3Bi(如图4d所示)。Bi的理论比容量为385 mAh/g,体积膨胀率为250%[115]。 Ellis等[115]对Bi的嵌钠行为进行了研究。他们发现在Bi的充放电曲线上存在两对平台,通过原位XRD分析可知两对平台对应的是NaBi相和Na3Bi相的生成。Su等[124]将Bi与石墨烯复合,在0.3~0.9 V电压范围内可逆比容量为358 mAh/g,在40 mA/g的电流密度下循环50周后,仍能保持大约200 mAh/g的可逆比容量。有趣的是,Su等通过XRD分析并没有发现Ellis等人所说的NaBi相和Na3Bi相。同时作者也注意到(012)晶面的峰位置随嵌入钠离子向右偏移,而脱出钠离子向左偏移,这种情况在后续的研究中也被观察到[125]。考虑到Bi是由褶皱的六元环组成的层状结构材料,其层间距为0.395 nm,完全可以容纳钠离子嵌入,因此作者认为Bi与Na反应不是发生合金化反应,而是嵌入式反应。后来的研究者也主要是将Bi和C复合[126, 127],或者制备成金属铋纳米棒阵列[125]以缓解Bi体积膨胀的问题。其中Yin等[127]利用静电纺丝的方法制备的一维Bi/C纳米纤维在100 mA/g的电流密度下,循环500周后仍然保持273.2 mAh/g的可逆比容量。最近,Wang等[128]直接以商业化的Bi作为电极,该电极在溶有1M NaPF6的二乙二醇二甲醚电解液中可逆比容量高达400 mAh/g,在400 mA/g的电流密度下循环2000周后容量保持率为94.4%,表现出高的储钠性能。虽然Bi有一定的储钠能力,但是在地壳中的含量不大,同时其理论嵌钠比容量只有385 mAh/g,与C基材料相比没有明显优势。但其具有较高的嵌入电势(相对于金属钠电位),可以考虑在高安全的储钠体系中使用。 根据计算,Ge也可以和Na形成NaGe合金,理论比容量为369 mAh/g,体积膨胀率为200%[129]。Baggetto等[130]制备的Ge薄膜可逆比容量为350 mAh/g,非常接近其理论容量,XRD结果显示Ge薄膜在完全脱钠和完全嵌钠的情况下呈现无定型态。Abel等[131]制备的纳米柱状Ge材料首周可逆比容量高达430 mAh/g,循环100周后容量保持率为88%,这高于Chevrier等人计算的理论比容量。接着Kohandehghan[132]和Lu[133]等人制备的Ge基材料的可逆比容量均超过了Chevrier计算的理论比容量,这说明Ge的理论比容量远远不止369 mAh/g。目前大家对Ge基材料的了解还比较有限,需要进一步的探讨研究。 合金类储钠负极拥有较高的比容量,其中Sn、Sb和P这3种材料拥有较高的理论比容量,是一类很有应用前景的储钠负极材料。合金类储钠负极材料的主要问题是在循环过程中面临着严重的体积效应,研究者们通过碳包覆、合金化和微纳结构设计等手段已经改进了合金类储钠负极的电化学性能。其中,碳包覆能有效提高合金材料的循环性能,合成过程简单,但是通常复合材料中碳所占比例较高,会降低整个电极的比容量,而失去合金负极高比容量的优势;合金化也能在一定程度上改善合金类储钠负极的循环性能,特别是与具有嵌钠电化学活性的金属形成的复合合金能够发挥各自储钠的优势,然而目前合金化对材料循环性能的提升仍有限,还需要结合碳包覆等方法进一步提升其电化学性能;通过微纳结构设计得到的材料具有有效缓冲体积变化和高比表面的优势,具有高的比容量和优异的循环与倍率性能,但是这类材料的制备过程较为复杂,成本较高,难以工业化生产。另外,微纳结构材料高的比表面也造成其首周充放电效率较低,这也是应用中需要考虑的一个问题。虽然上述方法能够大大提高合金类储钠负极的电化学性能,然而大部分材料的循环性能仍需改善,同时很少有研究者关注材料的首周库伦效率,而首周库伦效率低会过多消耗电池中有限的钠离子,造成电池容量下降。因此,更好的循环性能、更高的首周库伦效率以及更简单高效的合成方法是未来合金类储钠负极的发展方向。 References [1] Zhang Yu(张 宇), Yu Guoqin(俞国勤), Shi Mingrong(施明融),etal.EastChinaElectricPower(华东电力) [J], 2008, 36(4) :91-93. [2] Yu Enke(俞恩科), Chen Liangjin(陈梁金).ZhejiangElectricPower(浙江电力)[J], 2011, 12: 4-8. [3] Muňoz-Márquez M, Saurel D, Gómez-Cámer J L,etal.AdvancedEnergyMaterials[J], 2017: 1700463. [4] Dahbi M, Yabuuchi N, Kubota K,etal.PhysicalChemistryChemicalPhysics[J], 2014, 16(29): 15007-15028. [5] Slater M D, Kim D, Lee E,etal.AdvancedFunctionalMaterials[J], 2013, 23(8): 947-958. [6] Kim H, Kim H, Ding Z,etal.AdvancedEnergyMaterials[J], 2016, 6(19): 1600943. [7] Sangster J, Pelton A D.JournalofPhaseEquilibria[J], 1993, 14(2): 250-255. [8] Park C M, Kim J H, Kim H,etal.ChemicalSocietyReviews[J], 2010, 39(8): 3115-3141. [9] Qian J, Chen Y, Wu L,etal.ChemicalCommunications[J], 2012, 48(56): 7070-7072. [10] Darwiche A, Marino C, Sougrati M T,etal.JournaloftheAmericanChemicalSociety[J], 2012, 134(51): 20805-20811. [11] Baggetto L, Ganesh P, Sun C N,etal.JournalofMaterialsChemistryA[J], 2013, 1(27): 7985. [12] Baggetto L, Hah H Y, Jumas J C,etal.JournalofPowerSources[J], 2014, 267: 329-336. [13] Allan P K, Griffin J M, Darwiche A,etal.JournaloftheAmericanChemicalSociety[J], 2016, 138(7): 2352-2365. [14] Zhu Y, Han X, Xu Y,etal.ACSNano[J], 2013, 7(7): 6378-6386. [15] Hou H, Jing M, Yang Y,etal.JournalofPowerSources[J], 2015, 284: 227-235. [16] Wu L, Lu H, Xiao L,etal.JournalofMaterialsChemistryA[J], 2015, 3(10): 5708-5713. [17] Zhang N, Liu Y, Lu Y,etal.NanoResearch[J], 2015, 8(10): 3384-3393. [18] Qiu S, Wu X, Xiao L,etal.ACSAppliedMaterials&Interfaces[J], 2016, 8(2): 1337-1343. [19] Ding Y L, Wu C, Kopold P,etal.Small[J], 2015, 11(45): 6026-6035. [20] Yang C, Li W, Yang Z,etal.NanoEnergy[J], 2015, 18: 12-19. [21] Nithya C, Gopukumar S.JournalofMaterialsChemistryA[J], 2014, 2(27): 10516-10525. [22] Hu L, Zhu X, Du Y,etal.ChemistryofMaterials[J], 2015, 27(23): 8138-8145. [23] Wu L, Pei F, Mao R,etal.ElectrochimicaActa[J], 2013, 87: 41-45. [24] Liu J, Yu L, Wu C,etal.NanoLetters[J], 2017, 17(3): 2034-2042. [25] Wu L, Hu X, Qian J,etal.Energy&EnvironmentalScience[J], 2014, 7(1): 323-328. [26] Luo W, Zhang P, Wang X,etal.JournalofPowerSources[J], 2016, 304: 340-345. [27] Baggetto L, Allcorn E, Unocic R R,etal.JournalofMaterialsChemistryA[J], 2013, 1(37): 11163. [28] Baggetto L, Marszewski M, Górka J,etal.JournalofPowerSources[J], 2013, 243: 699-705. [29] Kim I T, Allcorn E, Manthiram A.EnergyTechnology[J], 2013, 1(5-6): 319-326. [30] Allcorn E, Manthiram A.ACSAppliedMaterials&Interfaces[J], 2014, 6(14): 10886-10891. [31] Baggetto L, Hah H Y, Johnson C E,etal.PhysicalChemistryChemicalPhysics[J], 2014, 16(20): 9538-9545. [32] Liu J, Yang Z, Wang J,etal.NanoEnergy[J], 2015, 16: 389-398. [33] Baggetto L, Allcorn E, Manthiram A,etal.ElectrochemistryCommunications[J], 2013, 27: 168-171. [34] Nam D H, Hong K S, Lim S J,etal.JournalofPowerSources[J], 2014, 247: 423-427. [35] Jackson E D, Green S, Prieto A L.ACSAppliedMaterials&Interfaces[J], 2015, 7(14): 7447-7450. [36] Liao S, Sun Y, Wang J,etal.ElectrochimicaActa[J], 2016, 211: 11-17. [37] Darwiche A, Toiron M, Sougrati M T,etal.JournalofPowerSources[J], 2015, 280: 588-592. [38] Darwiche A, Sougrati M T, Fraisse B,etal.ElectrochemistryCommunications[J], 2013, 32: 18-21. [39] Ji L, Zhou W, Chabot V,etal.ACSAppliedMaterials&Interfaces[J], 2015, 7(44): 24895-24901. [40] Walter M, Doswald S, Kovalenko M V.JournalofMaterialsChemistryA[J], 2016, 4(18): 7053-7059. [41] Xiao L, Cao Y, Xiao J,etal.ChemicalCommunications[J], 2012, 48(27): 3321-3323. [42] Farbod B, Cui K, Kalisvaart W P,etal.ACSNano[J], 2014, 8(5): 4415-4429. [43] Zhao Y, Manthiram A.ChemistryofMaterials[J], 2015, 27(8): 3096-3101. [44] Zhang W, Mao J, Pang W K,etal.ElectrochimicaActa[J], 2017, 235: 107-113. [45] Xie H, Kalisvaart W P, Olsen B C,etal.JournalofMaterialsChemistryA[J], 2017, 5(20): 9661-9670. [46] He M, Kravchyk K, Walter M,etal.NanoLetters[J], 2014, 14(3): 1255-1262. [47] Hou H, Jing M, Yang Y,etal.ACSAppliedMaterials&Interfaces[J], 2014, 6(18): 16189-16196. [48] Hou H, Jing M, Yang Y,etal.JournalofMaterialsChemistryA[J], 2015, 3(6): 2971-2977. [49] Hou H, Jing M, Zhang Y,etal.JournalofMaterialsChemistryA[J], 2015, 3(34): 17549-17552. [50] Liang L, Xu Y, Wang C,etal.Energy&EnvironmentalScience[J], 2015, 8(10): 2954-2962. [51] Liu S, Feng J, Bian X,etal.Energy&EnvironmentalScience[J], 2016, 9(4): 1229-1236. [52] Gu J, Du Z, Zhang C,etal.AdvancedEnergyMaterials[J], 2017: 1700447. [53] Ellis L D, Hatchard T D, Obrovac M N.JournaloftheElectrochemicalSociety[J], 2012, 159(11): A1801-A1805. [54] Chevrier V L, Ceder G.JournaloftheElectrochemicalSociety[J], 2011, 158(9): A1011-A1014. [55] Komaba S, Matsuura Y, Ishikawa T,etal.ElectrochemistryCommunications[J], 2012, 21: 65-68. [56] Wang J W, Liu X H, Mao S X,etal.NanoLetters[J], 2012, 12(11): 5897-5902. [57] Baggetto L, Ganesh P, Meisner R P,etal.JournalofPowerSources[J], 2013, 234: 48-59. [58] Baggetto L, Bridges C A, Jumas J C,etal.JournalofMaterialsChemistryA[J], 2014, 2(44): 18959-18973. [59] Du Z, Dunlap R A, Obrovac M N.JournalofAlloysandCompounds[J], 2014, 617: 271-276. [60] Stratford J M, Mayo M, Allan P K,etal.JournaloftheAmericanChemicalSociety[J], 2017, 139(21): 7273-7286. [61] Zhu H, Jia Z, Chen Y,etal.NanoLetters[J], 2013, 13(7): 3093-3100. [62] Liu Y, Zhang N, Jiao L,etal.AdvancedMaterials[J], 2015, 27(42): 6702-6707. [63] Mao M, Yan F, Cui C,etal.NanoLetters[J], 2017, 17(6): 3830-3836. [64] Chen W, Deng D.Carbon[J], 2015, 87: 70-77. [65] Liu Y, Zhang N, Jiao L,etal.AdvancedFunctionalMaterials[J], 2015, 25(2): 214-220. [66] Li S, Wang Z, Liu J,etal.ACSAppliedMaterials&Interfaces[J], 2016, 8(30): 19438-19445. [67] Luo B, Qiu T, Ye D,etal.NanoEnergy[J], 2016, 22: 232-240. [68] Pan F, Zhang W, Ma J,etal.ElectrochimicaActa[J], 2016, 196: 572-578. [69] Ellis L D, Ferguson P P, Obrovac M N.JournaloftheElectrochemicalSociety[J], 2013, 160(6): A869-A872. [70] Zhu J, Deng D.TheJournalofPhysicalChemistryC[J], 2015, 119(37): 21323-21328. [71] Lin Y M, Abel P R, Gupta A,etal.ACSAppliedMaterials&Interfaces[J], 2013, 5(17): 8273-8277. [72] Kim J C, Kim D W.Chemistry-AnAsianJournal[J], 2014, 9(11): 3313-3318. [73] Kim I T, Allcorn E, Manthiram A.JournalofPowerSources[J], 2015, 281: 11-17. [74] Kim Y, Kim Y, Choi A,etal.AdvancedMaterials[J], 2014, 26(24): 4139-4144. [75] Li W, Chou S L, Wang J Z,etal.AdvancedMaterials[J], 2014, 26(24): 4037-4042. [76] Qian J, Xiong Y, Cao Y,etal.NanoLetters[J], 2014, 14(4): 1865-1869. [77] Liu J, Kopold P, Wu C,etal.Energy&EnvironmentalScience[J], 2015, 8(12): 3531-3538. [78] Li Q, Li Z, Zhang Z,etal.AdvancedEnergyMaterials[J], 2016, 6(15): 1600376. [79] Shin H S, Jung K N, Jo Y N,etal.ScientificReports[J], 2016, 6: 26195. [80] Lan D, Wang W, Shi L,etal.JournalofMaterialsChemistryA[J], 2017, 5(12): 5791-5796. [81] Liu J, Wen Y, Van Aken P A,etal.NanoLetters[J], 2014, 14(11): 6387-6392. [82] Abel P R, Fields M G, Heller A,etal.ACSAppliedMaterials&Interfaces[J], 2014, 6(18): 15860-15867. [83] Xin F X, Tian H J, Wang X L,etal.ACSAppliedMaterials&Interfaces[J], 2015, 7(15): 7912-7919. [84] Vogt L O, Villevieille C.JournaloftheElectrochemicalSociety[J], 2016, 163(7): A1306-A1310. [85] Yamamoto T, Nohira T, Hagiwara R,etal.ElectrochimicaActa[J], 2016, 211: 234-244. [86] Vogt L O, Villevieille C.JournalofMaterialsChemistryA[J], 2016, 4(48): 19116-19122. [87] Nam D H, Kim T H, Hong K S,etal.ACSNano[J], 2014, 8(11): 11824-11835. [88] Kim C, Lee K Y, Kim I,etal.JournalofPowerSources[J], 2016, 317: 153-158. [89] Yabuuchi N, Kubota K, Dahbi M,etal.ChemicalReviews[J], 2014, 114(23): 11636-11682. [90] Sun J, Lee H W, Pasta M,etal.NatureNanotechnology[J], 2015, 10(11): 980-985. [91] Kim Y, Park Y, Choi A,etal.AdvancedMaterials[J], 2013, 25(22): 3045-3049. [92] Li W J, Chou S L, Wang J Z,etal.NanoLetters[J], 2013, 13(11): 5480-5484. [93] Qian J, Wu X, Cao Y,etal.AngewandteChemieInternationalEdition[J], 2013, 52(17): 4633-4636. [94] Hembram K P S S, Jung H, Yeo B C,etal.TheJournalofPhysicalChemistryC[J], 2015, 119(27): 15041-15046. [95] Hembram K P, Jung H, Yeo B C,etal.PhysicalChemistryChemicalPhysics[J], 2016, 18(31): 21391-21397. [96] Zhang M, Hu R, Liu J,etal.ElectrochemistryCommunications[J], 2017, 77: 85-88. [97] Li W, Ke L, Wei Y,etal.JournalofMaterialsChemistryA[J], 2017, 5(9): 4413-4420. [98] Yang Q R, Li W J, Chou S L,etal.RSCAdvances[J], 2015, 5(98): 80536-80541. [99] Li W J, Chou S L, Wang J Z,etal.ChemicalCommunications[J], 2015, 51(17): 3682-3685. [100] Li W J, Yang Q R, Chou S L,etal.JournalofPowerSources[J], 2015, 294: 627-632. [101] Ge X, Li Z, Yin L.NanoEnergy[J], 2017, 32: 117-124. [102] Pei L, Zhao Q, Chen C,etal.ChemElectroChem[J], 2015, 2(11): 1652-1655. [103] Ding X, Huang Y, Li G,etal.ElectrochimicaActa[J], 2017, 235: 150-157. [104] Lee G H, Jo M R, Zhang K,etal.JournalofMaterialsChemistryA[J], 2017, 5(7): 3683-3690. [105] Zhu Y, Wen Y, Fan X,etal.ACSNano[J], 2015, 9(3): 3254-3264. [106] Ruan B, Wang J, Shi D,etal.JournalofMaterialsChemistryA[J], 2015, 3(37): 19011-19017. [107] Li W, Hu S, Luo X,etal.AdvancedMaterials[J], 2017, 29(16): 1605820. [108] Marino C, Debenedetti A, Fraisse B,etal.ElectrochemistryCommunications[J], 2011, 13(4): 346-349. [109] Liu Y, Zhang A, Shen C,etal.ACSNano[J], 2017, 11(6): 5530-5537. [110] Zhou J, Liu X, Cai W,etal.AdvancedMaterials[J], 2017: 1700214. [111] Ren X, Lian P, Xie D,etal.JournalofMaterialsScience[J], 2017, 52(17): 10364-10386. [112] Huang Z, Hou H, Zhang Y,etal.AdvancedMaterials[J], 2017: 1702372. [113] Liu S, Feng J, Bian X,etal.Energy&EnvironmentalScience[J], 2017, 10(5): 1222-1233. [114] Yabuuchi N, Matsuura Y, Ishikawa T,etal.ChemElectroChem[J], 2014, 1(3): 580-589. [115] Ellis L D, Wilkes B N, Hatchard T D,etal.JournaloftheElectrochemicalSociety[J], 2014, 161(3): A416-A421. [116] Jow T R, Shacklette L W, Maxfield M,etal.JournaloftheElectrochemicalSociety[J], 1987, 134(7): 1730-1733. [117] Tran T T, Obrovac M N.JournalofTheElectrochemicalSociety[J], 2011, 158(12): A1411-A1416. [118] Sangster J, Pelton A D.JournalofPhaseEquilibria[J], 1992, 13(1): 67. [119] Malyi O I, Tan T L, Manzhos S.AppliedPhysicsExpress[J], 2013, 6(2): 027301. [120] Kim Y, Ha K H, Oh S M,etal.Chemistry-AEuropeanJournal[J], 2014, 20(38): 11980-11992. [121] Jung S C, Jung D S, Choi J W,etal.TheJournalofPhysicalChemistryLetters[J], 2014, 5(7): 1283-1288. [122] Xu Y, Swaans E, Basak S,etal.AdvancedEnergyMaterials[J], 2016, 6(2): 1501436. [123] Lim C H, Huang T Y, Shao P S,etal.ElectrochimicaActa[J], 2016, 211: 265-272. [124] Su D, Dou S, Wang G.NanoEnergy[J], 2015, 12: 88-95. [125] Liu S, Feng J, Bian X,etal.JournalofMaterialsChemistryA[J], 2016, 4(26): 10098-10104. [126] Yang F, Yu F, Zhang Z,etal.Chemistry-AEuropeanJournal[J], 2016, 22(7): 2333-2338. [127] Yin H, Li Q, Cao M,etal.NanoResearch[J], 2017, 10(6): 2156-2167. [128] Wang C, Wang L, Li F,etal.AdvancedMaterials[J], 2017, 29(35): 1702212. [129] Chou C Y, Lee M, Hwang G S.TheJournalofPhysicalChemistryC[J], 2015, 119(27): 14843-14850. [130] Baggetto L, Keum J K, Browning J F,etal.ElectrochemistryCommunications[J], 2013, 34: 41-44. [131] Abel P R, Lin Y M, De Souza T,etal.TheJournalofPhysicalChemistryC[J], 2013, 117(37): 18885-18890. [132] Kohandehghan A, Cui K, Kupsta M,etal.NanoLetters[J], 2014, 14(10): 5873-5882. [133] Lu X, Adkins E R, He Y,etal.ChemistryofMaterials[J], 2016, 28(4): 1236-1242. Research Progress on Alloy Anode Materials for Sodium Ion Batteries LIU Chuang,LU Haiyan,CAO Yuliang (College of Chemistry and Molecular Science, Wuhan University, Wuhan 430072, China) Sodium-ion batteries (SIBs) have attracted wide attention in the field of large-scale energy storage systems because of the extensive sources and low cost of sodium compounds. Anode materials have a great influence on the performance of batteries. Amongst various anode materials, alloy anode materials have caused extensive concern due to high capacity. However, the poor cycling stability resulting from severe volume expansion during charging and discharging hinders the development of alloy anode materials. Recently, the cycling stability of alloy materials has been greatly enhanced by coating, nanosizing or intermetallics. In this paper, we briefly review the reaction mechanism and research progress of Sn, Sb, P, Ge, Bi, Pb and Si materials alloying with sodium, discuss the problems of the alloy materials and the ways to solve them. Finally, we prospect the future development tendency of alloy anode materials for sodium-ion batteries. sodium ion battery; anode; alloy; mechanism; antimony; tin; phosphorus TM912 A 1674-3962(2017)10-0718-10 (编辑 惠 琼)2.2 Sn基材料
2.3 P基材料
2.4 Pb基材料

2.5 Si基材料
2.6 Bi基材料
2.7 Ge基材料
3 结 语
猜你喜欢
中国自行车(2022年5期)2022-09-06
蓄电池(2022年1期)2022-02-25
汽车工程师(2021年12期)2022-01-18
昆虫学报(2021年7期)2021-08-18
陶瓷学报(2020年3期)2020-10-27
黑龙江工业学院学报(综合版)(2020年8期)2020-10-23
电动工具(2020年2期)2020-04-22
科技创新与应用(2017年11期)2017-04-27
中华老年多器官疾病杂志(2016年2期)2016-01-16
科技资讯(2015年8期)2015-07-02
