
新型帕唑帕尼衍生物的合成与活性初步研究
2017-12-26 01:16李立威
华中师范大学学报(自然科学版) 2017年6期
刘 娥, 李立威
(1.荆楚理工学院 化工与药学院, 湖北 荆门 448000;2.荆楚理工学院 药物合成与优化湖北省重点实验室, 湖北 荆门 448000)
新型帕唑帕尼衍生物的合成与活性初步研究
刘 娥1,2*, 李立威1
(1.荆楚理工学院 化工与药学院, 湖北 荆门 448000;2.荆楚理工学院 药物合成与优化湖北省重点实验室, 湖北 荆门 448000)
在研究帕唑帕尼构效关系的基础上,以3-甲基-6-硝基吲唑为原料,设计合成4个帕唑帕尼衍生物.化合物结构经过1H NMR、LC-MS及元素分析确证,并进行体外抗肿瘤细胞增殖活性研究.采用四氮唑盐(MTT)法测试了4个化合物对MGC803细胞增殖的抑制作用.结果表明,目标化合物对于MGC803细胞增殖均有一定的抑制作用.
抗肿瘤活性; 酪氨酸激酶抑制剂; 合成; 帕唑帕尼衍生物
帕唑帕尼是一种多靶点酪氨酸激酶抑制剂,由英国葛兰素史克(GSK)公司开发.该药通过抑制血管内皮生长因子(VEGFR)阻断VEGF信号在肿瘤细胞中的表达,进而抑制血管的生成,达到“饿死”肿瘤的目的.帕唑帕尼作为血管肿瘤靶向药物,具有以下优点:不易产生耐药性,正常组织血管不易受到损伤;口服生物利用度高,抗瘤谱广.该药于2009年经美国食品药品管理局(FDA)批准上市,用于治疗晚期肾癌[1-4],2012年FDA又批准用于治疗进展性软组织肉瘤.目前,又联合其他药物开展其他癌症的治疗[5],如:卵巢癌、非小细胞肺癌等.尽管帕唑帕尼在临床上表现出较为理想的有效性和安全性,但其同时仍然存在一些不良反应[6],如:高血压、恶心、呕吐、腹泻、毛发颜色改变等.因此,为了降低其副作用,增强疗效,本课题组在研究帕唑帕尼的构效关系的基础上,参考相关文献[7-9],设计、合成了4个帕唑帕尼的衍生物,结构经过1H NMR和LC-MS确证,并初步对其体外抗肿瘤细胞增殖活性进行了评价[10-11].其合成路线及结构见图1.
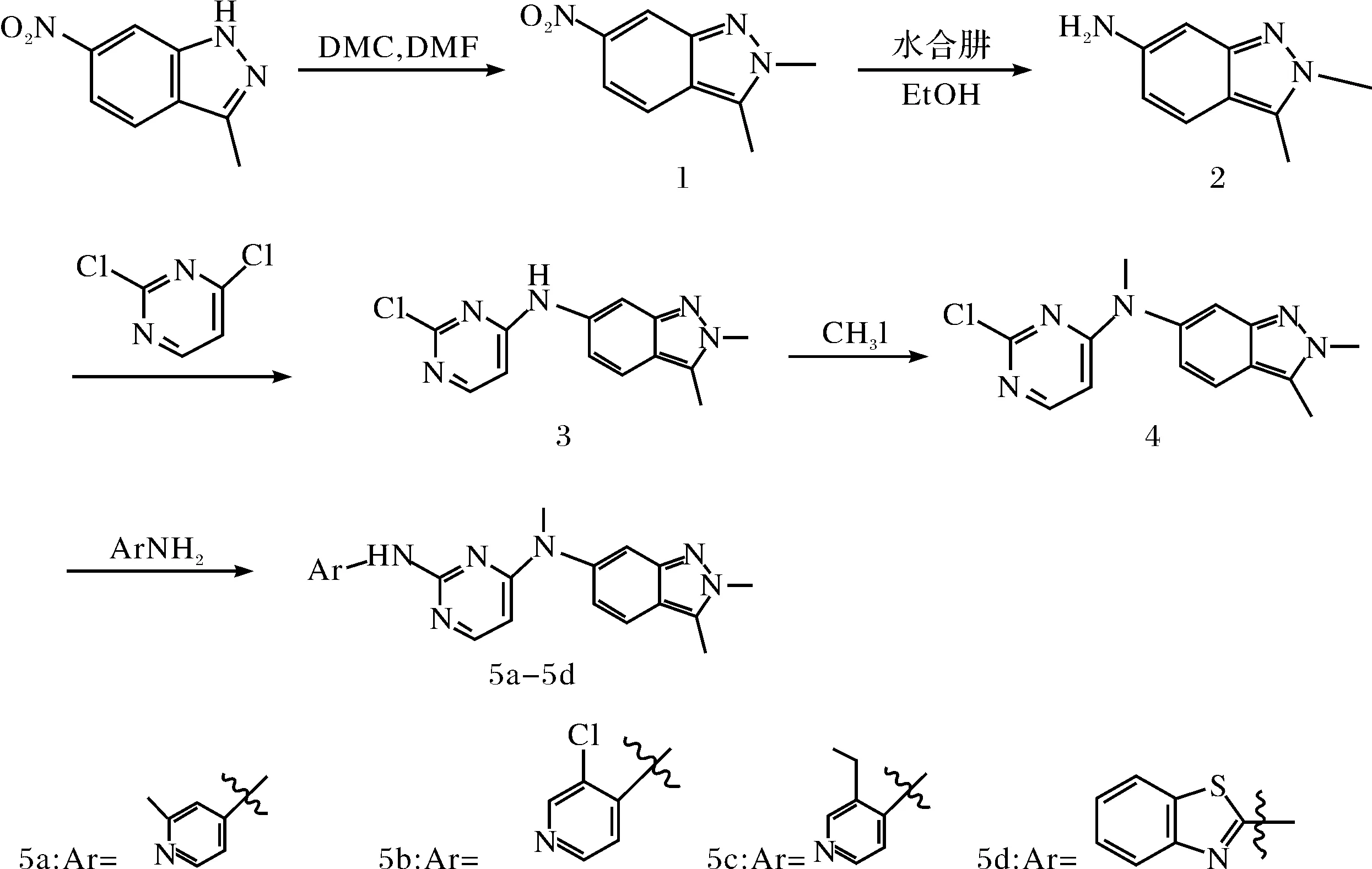
图1 目标化合物的结构及合成路线Fig.1 Structure and synthesis of target compounds
1 实验部分
1.1 试剂与仪器
DPX-400型核磁共振仪;UltiMate3000型液相色谱仪,HPLC-MS/MS (Thermo LCQFleet ),配备电喷雾离子化源(ESI).YRT-3型熔点仪(温度计未经校正),天津天大天发科技有限公司;RE-5298型旋转蒸发仪,上海亚荣生化仪器厂;Vario EL cube型元素分析仪,德国元素公司.
帕唑帕尼为实验室自制,其余试剂为国产分析纯或化学纯.
1.2 实验方法
1.2.1 中间体2,3-二甲基-6-硝基吲唑(1)的合成 将3-甲基-6-硝基吲唑(7.08 g,40 mmol)加入到装有50 mL DMF的烧瓶中,随后加入碳酸二甲酯(7.2 g,80 mmol),加完后,将反应液加热至100℃,搅拌约12 h,通过TLC检测反应终点.停止反应后,待反应液冷却至室温,将其倒入500 mL冰水中,搅拌,用饱和碳酸氢钠溶液调pH=9,过滤,得粗品.粗品经柱层析分离得黄色固体(化合物1).收率为80.3%,m.p.183.2~184.6℃.1H NMR(400MHz, CDCl3),δ:2.69(s,3H), 4.22(s,3H), 7.65(d,1H), 7.85 (d,1H), 8.61(s,1H);ESI-MS(m/z):192.2 [M+H]+.
1.2.2 中间体2,3-二甲基-6-氨基吲唑(2)的合成 将化合物1(5.65 g,0.03 mol)溶于50 mL无水乙醇中,缓慢加入20 mL水合肼,后加入1 g FeCl3,0.5 g活性炭,加热回流4 h,TLC监测反应,停止反应后,趁热过滤,滤液用饱和碳酸氢钠调至pH=9左右,用二氯甲烷萃取(50 mL×3),合并有机相,用无水硫酸镁干燥,蒸干,得浅黄色固体(化合物2),收率为91.5%,1H NMR(400MHz, DMSO-d6),δ: 2.65 (s, 3H), 3.85(s, 2H), 4.06 (s, 3H), 7.21(s, 1H), 7.74(d, 1H), 7.83(m, 1H); ESI-MS(m/z):162.3 [M+H]+.
1.2.3 中间体N-(2-氯嘧啶-4-基)-2,3-二甲基吲唑-6胺(3)的合成 将化合物2(4.83 g,0.03 mol)溶于40 mL无水乙醇中,加入碳酸氢钠3.8 g(0.045 mol),然后加入2,4-二氯嘧啶8.9 g(0.06 mol),加热回流4 h,反应结束后,冷却至室温,用无水乙醇洗,得白色固体(化合物3),收率为92.3%, m.p.215.8~216.6℃,1H NMR(400 MHz, DMSO-d6),δ:2.45(s, 3H), 3.87(s, 3H), 6.51(d, 1H), 6.84(m, 1H), 7.27(s, 1H), 7.59(t, 2H), 8.11(d, 1H); ESI -MS(m/z):274.1 [M+H]+.
1.2.4 中间体N-(2-氯嘧啶-4-基)-N,2,3-三甲基吲唑-6胺(4)的合成 在250 mL烧瓶中加入化合物3(5.46 g,0.02 mol),加入DMF100 mL使之溶解,后加入碳酸铯9.8 g(0.03 mol)和碘甲烷4.2 g(0.03 mol),室温下搅拌1 h,后将反应液倒入冰水中,搅拌使固体充分析出,抽滤,水洗得淡黄色固体(化合物4),收率为82.7%, m.p.175.3~177.2℃,1H NMR(400 MHz, DMSO-d6),δ:2.66(s, 3H), 3.45(s, 3H), 4.05(s, 3H), 7.29(d, 1H), 7.48 (d, 1H), 7.72(d, 1H), 7.86(d, 1H), 7.92(s, 1H); ESI-MS(m/z):288.2 [M+H]+.
1.2.5 化合物5a~5d的制备 在250 mL烧瓶中加入化合物4(5.75 g,0.02 mol)和50 mL无水乙醇,向其中加入相应的芳胺,搅拌下加热回流5 h,经TLC显示反应完全,冷却至室温,过滤,滤饼用乙醇淋洗(20 mL×3),得粗品,后经过柱层析纯化(乙酸乙酯∶石油醚=1∶5)得目标化合物5a~5d.
5a:白色固体,收率72.8%, m.p.:181.7~182.5℃,1H NMR(400 MHz, DMSO-d6),δ:2.20(s, 3H), 2.77(s, 3H), 3.33(s, 3H), 3.52(s, 3H), 5.68 (d, 1H), 6.33(d, 1H), 6.42(s, 1H), 6.58(d, 1H),6.69(s, 1H), 7.72(d, 1H), 7.56(d, 1H), 8.55(s, 1H), 9.13(s, 1H); ESI-MS(m/z):360.2 [M+H]+.元素分析(%): C 66.18, H 5.93, N 26.82, 理论值(%): C 66.82, H 5.84, N 27.30.
5b:白色固体,收率68.4%, m.p.:203.3~204.7℃,1H NMR(400 MHz, DMSO-d6),δ: 2.75(s, 3H), 3.37(s, 3H), 3.71(s, 3H), 5.70(d, 1H), 6.35(d, 1H), 6.63(s, 1H), 7.20(s, 1H), 7.70(d, 1H),7.65(d, 1H), 8.66(s, 1H), 8.74(s, 1H), 9.15(s, 1H);ESI-MS(m/z):380.5 [M+H]+.元素分析(%): C 60.15, H 4.96, N 25.13, 理论值(%): C 60.08, H 4.74, N 25.82.
5c:类白色固体,收率76.7%, m.p.:194.7~195.9℃,1H NMR(400 MHz, DMSO-d6),δ: 1.35 (s, 3H), 2.54(t, 2H), 2.77(s, 3H), 3.35(s, 3H), 3.62 (s, 3H), 5.71(d, 1H), 6.41(d, 1H),6.70(s, 1H), 6.91(s, 1H), 7.31(s, 1H), 7.74(d, 1H), 8.35(s, 1H), 8.68(s, 1H), 9.36(s, 1H); ESI-MS(m/z):374.4 [M+H]+.元素分析(%): C 67.15, H 6.09, N 26.15, 理论值(%): C 67.56, H 6.17, N 26.27.
5d:白色固体,收率73.2%, m.p.:217.7~218.5℃,1H NMR(400 MHz, DMSO-d6),δ: 2.56(s, 3H), 3.21(s, 3H), 3.54 (s, 3H), 5.73(d, 1H), 6.40(s, 1H), 6.82 (s, 1H), 7.55 (s, 1H), 7.61 (s, 1H), 7.69(d, 1H), 7.77(d, 1H), 8.12(s, 1H), 8.37(s, 1H), 9.89(s, 1H); ESI-MS(m/z):390.7 [M+H]+.元素分析(%): C 61.21, H 4.35, N 24.97, S 8.06;理论值(%): C 61.70, H 4.88, N 25.19, S 8.23.
1.3 目标化合物对肿瘤细胞的抑制活性评价
采用四氮唑盐(MTT)法,以帕唑帕尼为阳性对照,将所得的4种目标化合物针对MGC803细胞进行体外抗肿瘤增殖活性评价.将处于对数生长期的细胞,用含10%胎牛血清的RPMI1640的培养液稀释至所需浓度,加入到96孔板中,培养24 h.受试化合物分别溶解并稀释成5个浓度(10-4,10-5,10-6,10-7,10-8),培养4~5 d,每孔加入10 mL 5 mg·mL-1的MTT溶液,培养4 h,吸出上清,加入200 μL的二甲亚砜使甲臜溶解,用酶标仪在570 nm处检测,并计算半数抑制浓度.结果显示,目标化合物5a~5d对MGC803细胞的IC50分别为(9.516±0.724)、(3.921±0.617)、(10.335±0.425)、(1.238±0.766)μmol·L-1, 阳性对照品帕唑帕尼的IC50为(4.925±0.537)μmol·L-1.其中5b和5d抑制活性略高于帕唑帕尼,可作为先导化合物进一步研究.
2 结果与讨论
2.1 合成部分
以3-甲基-6-硝基吲唑为起始原料,经过甲基化得到中间体2,3-二甲基-6-硝基吲唑,有文献报道该步采用硫酸二甲酯,但是考虑到硫酸二甲酯的毒性以及对环境的污染,笔者改用碳酸二甲酯进行甲基化.
在将硝基还原为氨基的过程中,文献一般采用Pd/C 催化加氢或Fe/NH4Cl还原,但考虑到成本以及后处理问题,本文采用了水合肼还原法,而且也获得了较好的收率.
设计了几个芳胺化合物,通过与中间体N-(2-氯嘧啶-4-基)-N,2,3-三甲基吲唑-6胺反应,制备得到目标化合物5a~5d,产物结构经过1H NMR、元素分析及LC-MS确证.
2.2 活性部分
采用MTT法对目标化合物进行了体外活性测试,结果表明:在嘧啶环上再引入吡啶环,均表现出一定的对MGC803细胞增殖的抑制作用,其中5b和5d抑制活性较好,从结构上来看,可能是因为在吡啶环上引入了卤素,另外,苯并噻唑基的引入也同样增加了化合物的抑制活性.
3 结论
本课题组在研究帕唑帕尼构效关系的基础上,以3-甲基-6-硝基吲唑为原料,设计合成了4个帕唑帕尼衍生物,目标化合物结构经过1H NMR、元素分析和LC-MS确证,对中间体的合成工艺也进行了改进.同时,以帕唑帕尼为阳性对照品,采用MTT法对目标化合物进行了体外抗肿瘤细胞增殖活性评价,发现目标化合物均有一定的抑制肿瘤细胞增殖的活性,其中化合物5b和5d有较好的抑制活性,可作为先导化合物进一步研究.
[1] JIA Y, ZHANG J, FENG J, et al. Design, synthesis and biological evaluation of pazopanib derivatives as antitumor agents[J]. Chem Biol Drug Des, 2014,83: 306-316.
[2] 吴智鸿, 赵砚瑾, 王永珍, 等. 帕唑帕尼衍生物的合成与初步的活性评价[J].化学工程师,2015,4:12-15.
[3] 吕水利, 王 鹏, 张艳利. 帕唑帕尼治疗晚期肾细胞癌的临床研究进展[J].中国医药指南,2013,12(11):340-341.
[4] PHILIP A H, AMOGH B, MUI C, et al. Discovery of 5-[[4-[(2,3-Dimethyl-2H-indazol-6-yl) methylamino]-2-pyrimidinyl]amino]-2-methylbenzenesulfonamide(Pazopanib), a novel and potent vascular endothelial growth factor receptor inhibitor[J]. J Med Chem, 2008,51: 4632-4640.
[5] GIROLAMO R, MARIA M, EUGENIO D D P,et al. Pazopanib a tyrosine kinase inhibitor with strong anti-angiogenetic activity: a new treatment for metastatic soft tissue sarcoma[J]. Critical Reviews in Oncology/Hematology,2014,89: 322-329.
[6] 郑宇静, 姜文亮, 王志宏, 等. 酪氨酸激酶抑制剂帕唑帕尼的药理与临床研究[J].中国新药杂志,2011,20(12):1057-1060.
[7] QI H, CHEN L, LIU B, et al. Synthesis and biological evaluation of novel pazopanib derivatives as antitumor agents[J]. Bioorganic & Medicinal Chemistry Letters, 2014,24: 1108-1110.
[8] 舒 婷, 李光兴. 碳酸二甲酯作甲基化试剂的研究进展[J].化工中间体, 2008(1) : 20-23.
[9] 刘梦露, 方 丽, 李振华. 盐酸帕唑帕尼的合成工艺改进[J].合成化学, 2014,22(1):121-123.
[10] 张晓凯, 刘登科, 刘冰妮, 等. 新型含嘧啶环吲唑衍生物的合成及初步生物活性研究[J].中国药物化学杂志, 2013,23(1):15-20.
[11] 李 伟, 雷春花, 王立强, 等. 多靶点受体酪氨酸激酶抑制剂喹啉衍生物的合成及活性初步研究[J].中国新药杂志, 2016,25(13):1522-1530.
Synthesisandpreliminaryanticanceractivityofnovelpazopanibderivatives
LIU E1,2, LI Liwei1
(1.College of Chemical Engineering and Pharmacy, Jingchu University of Technology, Jingmen, Hubei 448000,China; 2.Hubei Key Laboratory of Drug Synthesis and Optimization, Jingchu University of Technology, Jingmen, Hubei 448000, China)
Based on the structure-activity relationship of pazopanib, four novel pazopanib derivatives were synthesized with 3-methyl-6-nitroindazole as raw materials. The structures of the compounds were identified by1H NMR, LC-MS and elemental analyses, and the anti-proliferation activity of tumor cells were studied in vitro. MTT method was used to test the inhibition effect of proliferation activity of the target compounds on MGC803 cell. The results showed that the target compounds were able to inhibit the proliferation of MGC803 cells.
antitumor activity; tyrosine kinase inhibitor; synthesis; pazopanib derivatives
2017-04-17.
湖北省自然科学基金项目(2013CFA015);药物合成与优化湖北省重点实验室开放基金项目(OPP2015YB04);湖北省教育厅科研项目(Q20164303).
*通讯联系人. E-mail: liue2011@126.com.
10.19603/j.cnki.1000-1190.2017.06.010
1000-1190(2017)06-0782-04
O625.63
A
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29
昆明医科大学学报(2021年8期)2021-08-13
昆明医科大学学报(2021年3期)2021-07-22
化工管理(2021年7期)2021-05-13
汕头大学学报(自然科学版)(2020年4期)2020-12-14
农药科学与管理(2019年6期)2019-11-23
中成药(2017年5期)2017-06-13
中国农资(2016年1期)2016-12-01
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
股市动态分析(2015年12期)2015-09-10
