
凯氏定氮仪测定饲料中粗蛋白含量误差因素分析
2018-01-25 13:08丁立人
农业与技术 2018年21期
丁立人
摘 要:利用全自动凯氏定氮仪的工作原理,通过对各实验操作步骤中有可能产生误差的来源作了详细的分析,从而达到减小实验误差,提高检测准确性的目的。
关键词:凯氏定氮仪;蛋白质含量;误差;准确度
中图分类号:S-3 文献标识码:A DOI:10.11974/nyyjs.20181131006
饲料中粗蛋白质含量是饲料质量的一个重要指标,目前常用的检测方法是凯氏定氮法,其通过测定样品中总氮量来计算样品蛋白质含量,适用于各类样品中蛋白质含量的测定,国内外应用较为普遍,迄今为止仍是饲料中蛋白质测定的国标规定方法。
全自动凯氏定氮仪同传统的半微量凯氏蒸馏装置相比,全自动凯氏定氮仪的定氮原理与之相同,并具有省时、省力、准确性高等优点,因而在饲料行业得到广泛应用。但测定粗蛋白的步骤依然繁多,每个步骤、每个细节都有可能人为因素造成误差,从而表现为结果的偏低、偏高以及重复性不好等几种形式。本文以FOSS Kjeltec8400型全自动凯氏定氮仪为例,通过对其整个实验过程的整理,根据实验操作的先后顺序,对各步骤中误差产生的可能因素和来源进行分析,并提出相应的避免或降低误差的解决方法。
1 方法原理
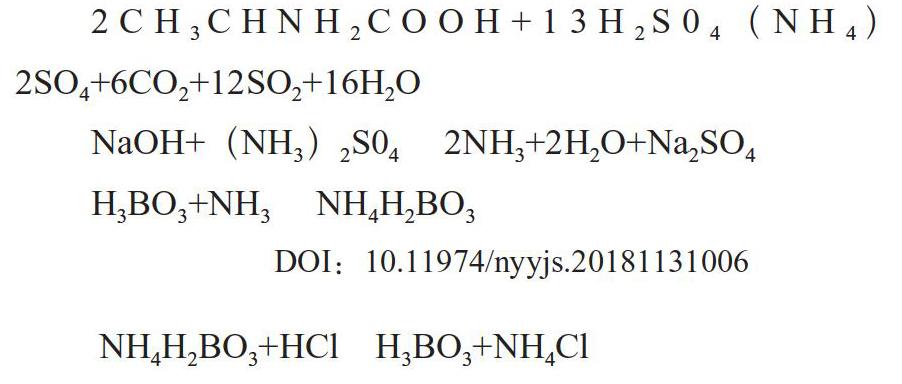
凯氏定氮法测定饲料中蛋白质的原理是各种饲料在催化剂(硫酸钾、硫酸铜)的作用下,用浓硫酸进行消化,使蛋白质和氨态氮都转变成氨气,并被浓硫酸吸收变为硫酸铵,而非含氮物质,则以二氧化碳、水、二氧化硫的氣体状态逸出。消化液在浓碱的作用下进行蒸馏,释放出的氨气随水蒸气蒸出并被硼酸液吸收生成硼酸胺,以甲基红-溴甲酚绿作混合指示剂,用盐酸标准液滴定,求出氮的含量,再乘以一定的换算系数(通常用6.25系数计算),得出样品中的粗蛋白质的含量。
其主要化学反应如下:
2CH3CHNH2COOH+13H2S04(NH4)2SO4+6CO2+12SO2+16H2O
NaOH+(NH3)2S04 2NH3+2H2O+Na2SO4
H3BO3+NH3 NH4H2BO3
NH4H2BO3+HCl H3BO3+NH4Cl
2 误差来源分析及解决措施
2.1 样品制备环节
2.1.1 样品的取样
测定要求样品粉碎并全部过40目筛,目的在于使其具备均质性,便于溶样,更易于消化。在粉碎或过筛过程中,样品容易产生自动分级,如玉米,其硬质颗粒大的部分常聚集在上面,粉质部分聚在下面。所以粉碎过筛后要重新混合均匀,以减少测量误差。
粉碎完后,粉碎机中总会遗留少部分,这部分不能随便丢弃,否则将影响样品的均质性造成测量误差。
对于全价料(即使是颗粒全价料)和浓缩料,无须再粉碎,当然这是基于混合机的良好混合均度的;如果拿去粉碎,会丢失部分粉末物质,且粉碎后产生分级,反而降低了样品的均质性,因此测定的样品一定要均匀,不均匀的样品会导致测定结果重复性差,结果或高或低。
2.1.2 样品的用量
在进行样品称量时2点很关键:样品的质量或体积的准确性以及最合适的上样量。
当固体样的称量质量高于实际质量或液体样品的量取体积大于实际的体积时, 就会导致最后定氮结果偏低。反之,则会导致定氮结果偏高。因此,称量用天平一定要定期校正,液体样品量取也要使用经过校正的移液器,以确保样品的质量或体积准确性。
样品的称取在0.5~1g左右比较适宜,对于鱼粉、豆粕、浓缩料等粗蛋白含量较高的样品,称量0.5g左右即可,而对于玉米、稻谷蛋白含量低的样品,适当增加称量至1g左右,如果取样太少,盐酸标准液滴定体积过小,增加实验误差,取样太多,消化时消耗的硫酸量大,消化不完全,导致结果偏低。同时,如果上样量超出仪器的检测范围也会导致定氮结果不准确。以FOSS Kjeltec 8400型全自动凯氏定氮仪为例,一般单次凯氏定氮检测的样品总含氮量应在 0.1~200mg。
2 .1.3 消化管清洁不彻底
消化管如果清洁不彻底可能会有样品残留,从而导致定氮结果偏高。所以,每次实验称量前要确保消化管清洗干净,称量纸应是无氮纸或者是定量滤纸,否则结果偏高。
样品放入消化管内时,注意不要粘附在管壁上,万一有粘附可用少量浓硫酸将其缓慢冲下,以免被检样消化不完全,结果偏低。
2.2 样品消煮环节
2.2.1 消化炉温度的控制
开始消化时消化炉要调低至200℃,待样品焦化,泡沫消失后,逐步缓慢加强火力,直至升到420℃,炉温过低消化时间长且不完全,过高会引起氮的损失,都会导致粗蛋白的结果偏低。水分含量高、糖分、油脂高的样品易起泡,可将催化剂和硫酸加入消化管内静置过夜再逐步升温消化,可在一定程度上减少发泡现象的产生,防止氮的损失。
2.2.2 硫酸與催化剂的比例
硫酸与催化剂的最佳比例为1.7(10mL硫酸,6g左右催化剂),提高沸点到385℃。比例不对,沸点达不到,消化不完全,结果偏低。
2.2.3 排废速率
排废的速率过高时,硫酸的消耗就会加快,参与消化的硫酸量会减少,从而使样品消化不完全而导致定氮结果偏低。因此在消化刚开始把抽气泵开到最大,约5~8min后关小抽气泵降低排废速度以致酸雾恰好充满排废罩保证硫酸回流,避免氨的损失。
2.2.4 消化时间
样品的消化时间对饲料中粗蛋白质测定也很重要,样品消化至透明蓝绿色液体再消化15min,说明消化完全,消化时间过短,饲料中的氮不能充分转化成氨;消化时间过长,酸消耗量过大,会造成氨的流失。
2.3 蒸馏环节
2.3.1 接收液的pH值
接收液为加指示剂的1%硼酸水溶液,正常颜色显示暗红色,如果溶液是红色,pH值偏酸,则应用5%氢氧化钠溶液调节至暗红色,反之接收液颜色偏绿,则需加5%盐酸溶液调至暗红,以确保接收液呈中性,避免接收液pH值不准确造成的结果误差。
2.3.2 蒸馏头变形或有异物
当蒸馏头上腐蚀变形或附着杂质固体异物,消化管与蒸馏头之间密封不严,就会引起氨气甚至液体泄漏,使定氮结果偏低。另外,多次实验之后由于清洗不彻底,蒸馏头上残留的有机氮杂质可能会在实验过程中计入正在蒸馏的样品当中,导致定氮结果偏高。因此,蒸馏头应定期用醋酸水溶液进行清洗,如发现蒸馏头腐蚀老化应更换蒸馏头。
2.3.3 氢氧化钠溶液的用量
在样品消化完全后,样品上机会加入浓碱液(一般为400g/L的氢氧化钠溶液 ) 使之呈碱性后进行加热蒸馏,使氨气释放。碱液的加入体积一般是硫酸体积的4倍以上,通常10~12mL硫酸消化,仪器程序设定50mLNaOH溶液,使反应液蒸馏过程保持褐色,说明碱足量,这样才能使氨完全游离,易于硼酸吸收。如果碱量不足时,会使得氨气释放不彻底从而引起最终定氮结果偏低,同时为了避免这种情况,并检查碱泵输送的体积是否正确,碱泵要定期用温水清洗,避免供碱系统堵塞。
2.3.4 蒸馏时间
当样品的含氮量较高而蒸馏的时间又过短时,蒸馏反应会不充分,氨气释放不完全,导致最终的定氮结果偏低。遇到这种情况应适当的延长蒸馏时间,确保样品的氨气充分释放;或者适当减少样品量,确保反应完全。蒸馏同时做好定氮仪系统密封性检查,保证接口密封不漏液,有利氨蒸馏。
2.4 滴定环节
2.4.1 盐酸标准液浓度的影响
盐酸标准液浓度是否准确尤为关键,如果标准酸的浓度偏高时,消耗标准酸的体积就会偏少,从而使最终的定氮结果偏低,反之则结果偏高。所以标准酸溶液标定时,增加平行标定的次数,可以减少随机误差。在一般分析工作中,测定次数为3~5次,如果没有意外误差发生,基本上可以得到比较准确的标定结果,同时妥善保存标液以防止浓度发生变化,影响定氮结果。
2.4.2 滴定系统中存在气泡
滴定系统中如果混有气泡,气泡占据标准盐酸的体积,使得计量的酸量就会大于实际量,从而导致结果偏高。为了避免这种情况,每次实验前要检查滴定系统,具体操作是采用手动排液和充满液体的方法将滴定系统内的气泡排出,并手动清洗干净滴定室,使得滴定室不再有残余的酸。
2.4.3 空白值的影响
空白值检测是衡量整个操作过程中一个非常重要的指标,良好的空白值是保证检测结果准确的前提。空白值偏高,那么在样品滴定时标准酸的实际消耗体积就会降低,从而导致最终的定氮结果偏低。引起空白值偏高的原因主要是消化管未清洗干净,或者是有污染物残留反应管道。所以,测定用的消化管要彻底清洁;蒸馏头应定期用醋酸水溶液进行清洗,确保没有有机氮的残留。
3 结语
应用全自动凯氏定氮仪测定饲料中的粗蛋白含量,大大提高了检测效率,而通过以上的分析,对全自动凯氏定氮仪测定粗蛋白可能的误差来源有个全面正确认识,并在日常测定中掌握正确的操作方法,避免每个环节人为因素的影响,一定能将误差控制在最小范围,提高测定样品粗蛋白含量的准确度。
猜你喜欢
哈尔滨轴承(2020年2期)2020-11-06
今日中国·法文版(2020年7期)2020-07-04
中国特种设备安全(2019年1期)2019-03-13
建筑科技(2018年6期)2018-08-30
中国交通信息化(2016年5期)2016-06-06
电测与仪表(2016年18期)2016-04-11
电测与仪表(2015年8期)2015-04-09
四川师范大学学报(自然科学版)(2015年1期)2015-02-28
天津冶金(2014年4期)2014-02-28
机电信息(2014年35期)2014-02-27
