
Development of a sensitive and reliable droplet digital PCR assay for the detection of ‘Candidatus Liberibacter asiaticus’
2018-02-05 07:11ZHONGXiLIUXueluLOUBinghaiZHOUChangyongWANGXuefeng
ZHONG Xi, LIU Xue-lu, LOU Bing-hai, ZHOU Chang-yong, WANG Xue-feng
1 National Citrus Engineering Research Center, Citrus Research Institute, Chinese Academy of Agricultural Sciences/Southwest University, Chongqing 400712, P.R.China
2 Guangxi Key Laboratory of Citrus Biology, Guangxi Academy of Specialty Crops, Guilin 541004, P.R.China
1. Introduction
‘CandidatusLiberibacter asiaticus’ (Las), a phloem-resided α-protebacterium, is the putative causal agent of citrus Huanglongbing (HLB, yellow shoot disease) that is one of the most serious diseases in citrus production (Bové 2006). The bacterium is transmitted from infected to healthy plants through grafting or by citrus psyllid (Diaphorina citri).No effective cure is currently available for HLB-infected citrus plants. Therefore, the use of pathogen-free nursery stocks, control of insect vector and removal of infected trees are major control measures in HLB management.This is of particular importance if HLB infection status of asymptomatic trees in field could be accurately diagnosed for the implementation of control strategies.
Because Las is unable to be cultured so far, current detection is typically PCR-based using primers developed from genomic DNA sequences, mostly the 16S rRNA gene.Primer set OI1/OI2c for conventional PCR and primer-probe set HLBas/HLBp/HLBr for TaqMan real-time quantitative PCR(qPCR) are widely used for the standardized detection of Las(Jagoueixet al. 1994; Liet al. 2006). Recently, multi-copy genes have been chosen as targets for the improvement of qPCR sensitivity (Morganet al. 2012; Zhenget al. 2016).However, absolute quantification of unculturable Las by qPCR is challenging due to erratic distribution and low titer,especially for early detection of Las infection.
Droplet digital PCR (ddPCR) is a new technology that allows sensitive detection and absolute quantification of low concentration DNA without the need for a standard curve.Each sample tested was partitioned in tens of thousands of individual droplets in a water-oil emulsion and then the number of positive droplets was read by cumulativefluorescence signal during PCR amplification. The total number of target DNA molecules in a sample can be calculated from the fraction of positive droplets and Poisson statistics (Hindsonet al. 2011). Since ddPCR has been shown to yield more precise detection results than qPCR,the robust and powerful method has been increasingly used in medical researches (Tayloret al. 2015), clinical applications (Tsuiet al. 2011; Watanabeet al. 2015), food safety inspection (Pinheiroet al. 2011; Florenet al. 2015)and gene-editing frequencies study (Mocket al. 2016).Recently, it also has been used to detectXanthomonas citrisubsp.citri, an economically important disease of citrus(Zhaoet al. 2016).
In this study, we established ddPCR approach to detect and quantify Las in both symptomatic and asymptomatic samples. The detection sensitivity of ddPCR was compared to qPCR targeting the gene encoding 16S rRNA.
2. Materials and methods
2.1. Sample collection and DNA extraction
HLB symptomatic and asymptomatic field citrus samples were collected from Guangxi and Hunan of China. All collected samples in China were shipped by mail to Citrus Research Institute (CRI) of Southwest University in Chongqing, China. Four HLB-positive citrus samples and four negative citrus samples were collected from the greenhouse in CRI. The midribs of citrus leaves were excised and DNA was extracted using the cetyltrimethylammonium bromide (CTAB) methods as previously described (Wanget al. 2012).
2.2. Preparation of cloned plasmid standard
A DNA segment encoding 16S rRNA gene of Las was amplified with Las genomic DNA as the template. The PCR amplicon was purified and ligated into the pEASY-T1 cloning vector (TransGen Biotech, China). Plasmid DNA was extracted from transformed competent cells and used to generate a standard curve for tenfold serial dilutions consisting of nine concentration gradients, which were used to test the sensitivities and linearity range of qPCR and ddPCR assays.
2.3. Quantitative PCR
The primers and probe targeted the 16S rRNA gene of Las were used in the subsequent qPCR and ddPCR assays (Liet al. 2006). The qPCR assay was performed on an iCyler IQTMSystem (Bio-Rad, Hercules, CA, USA). The cycling conditions included incubation for 30 s at 95°C followed by 40 cycles of 95°C for 5 s and 58°C for 30 s. Ctvalues were analyzed using BioRad iCycler iQ version 3.0 Software with auto-calculated baseline settings and a manually set threshold at 0.1. Standard curve was constructed through serial dilutions of plasmids for quantification and checked for qPCR efficiencies.
2.4. Droplet digital PCR
The QX200TMDroplet Digital PCR System (Bio-Rad,Hercules, CA, USA) was used in the study. The total ddPCR reaction volume was 20 μL, containing 10 μL 2× ddPCRTMsupermix for probe (no dUTP) (Bio-Rad, Pleasanton, CA,USA), 1 μmol L–1of each primer, 500 nmol L–1of probe, and 2 μL template DNA. Approximately 20 000 droplets were generated using a Droplet Generator (DG) with an 8-channel DG8 cartridge and cartridge holder with 70 μL of DG oil per well and 20 μL of reaction mixture. Following this step, 40 μL droplets mixtures were transferred into a 96-well plate. The PCR plate was heat-sealed using a PX1TMPCR Plate Sealer(Bio-Rad) and placed in the C1000 Thermal Cycler (Bio-Rad)under the following thermal conditions (temperature ramp rate 2°C s–1): 95°C for 10 min, followed by 40 cycles of 94°C for 30 s and 54°C for 1 min. Droplets were counted on the QX200 droplet reader (Bio-Rad).
2.5. Data analysis
Linear regression analyses of standard curve from qPCR was performed and recalculated with Microsoft Excel Software (Microsoft, USA). Slope value of standard curve was used to determine PCR efficiencies. For ddPCR,positive droplets were discriminated from negative droplets by applying a fluorescence amplitude threshold with the QuantaSoftTMversion 1.7.4 (Bio-Rad). Correlation analysis between ddPCR and qPCR was performed with SPSS Software version 21.0 (SPSS Inc., Chicago, USA).Pearson’s correlations and linear regression were also used to evaluate the relationship between measurements of ddPCR and qPCR assays.
3. Results and discussion
Adequate discrimination between positive and negative signals is of great importance to set appropriate thresholds.Annealing temperature conditions play important roles in determining fluorescence intensity and the distance between positive and negative signals. To assess the optimal annealing temperature of the ddPCR assay, the eight temperature gradients ranged from 64 to 52°C were set on the thermal cycler. An optimized annealing temperature of 54°C was determined based on the largest discrimination in fluorescence intensity between positive and negative droplets.
To compare the linearity, dynamic range and sensitivity of qPCR and ddPCR assays, calibration curves for the qPCR assay and the regression curves for the ddPCR assay were constructed using tenfold serial dilutions of positive plasmid(3.07×108–3.07×101copies µL–1). Both qPCR and ddPCR assays exhibited good linearity of amplification with high determination coefficient (R2=0.999 and 0.996, respectively)(Fig. 1-A and B). Furthermore, a very strong and significant positive correlation between the two methods (r=0.99;P<0.001) was observed (Fig. 1-C). The dynamic range tested in positive plasmid in qPCR was from 108to 102.Compared to qPCR, ddPCR had the narrower linearity range from 105to 101copies since the droplets were positively saturated at target concentrations ≥106copies μL–1, making the Poisson algorithm invalid (Fig. 2-A). However, ddPCR showed a lower detection limit, suggesting the ddPCR is more sensitive than qPCR (Fig. 2).
The weak real-time PCR signals derived from lowconcentration samples, as represented by high Ctvalues,may be questionable for declaring a positive reaction.To better compare the detection sensitivity between the two assays, samples with Ct>35 tested by qPCR were regarded as Las-negative samples in this study. Total of 40 citrus samples extracted previously, with the Ctvalue ranging from 28 to 38 by qPCR, were chosen for testing the detection capacity of ddPCR for high Ctvalues samples and determining whether ddPCR can be used in the detection of field samples. Besides the relatively low Ctvalue (<35)samples, six of 13 samples (46.15%) with high Ctvalue(>35) were also positive by ddPCR (data not shown). It should be noted that asymptomatic citrus samples with low Las concentration could be detected by ddPCR, suggesting that ddPCR is a more robust method for the detection of samples with low concentration of Las, especially for samples in early infection and asymptomatic phase. The application of a microsimulation model of asymptomatic disease spread using psyllid introduction scenarious indicated that the surveillance and control should be used from the initial detection of invasion and throughout the asymptomatic period (Leeet al. 2015). The ddPCR-based technology will play an important role in the detection of Las from asymptomatic citrus samples. It is believed that if more primer pairs targeting multi-copy genes were used in ddPCR (Morganet al. 2012; Zhenget al. 2016), the detection sensitivity might be improved accordingly.
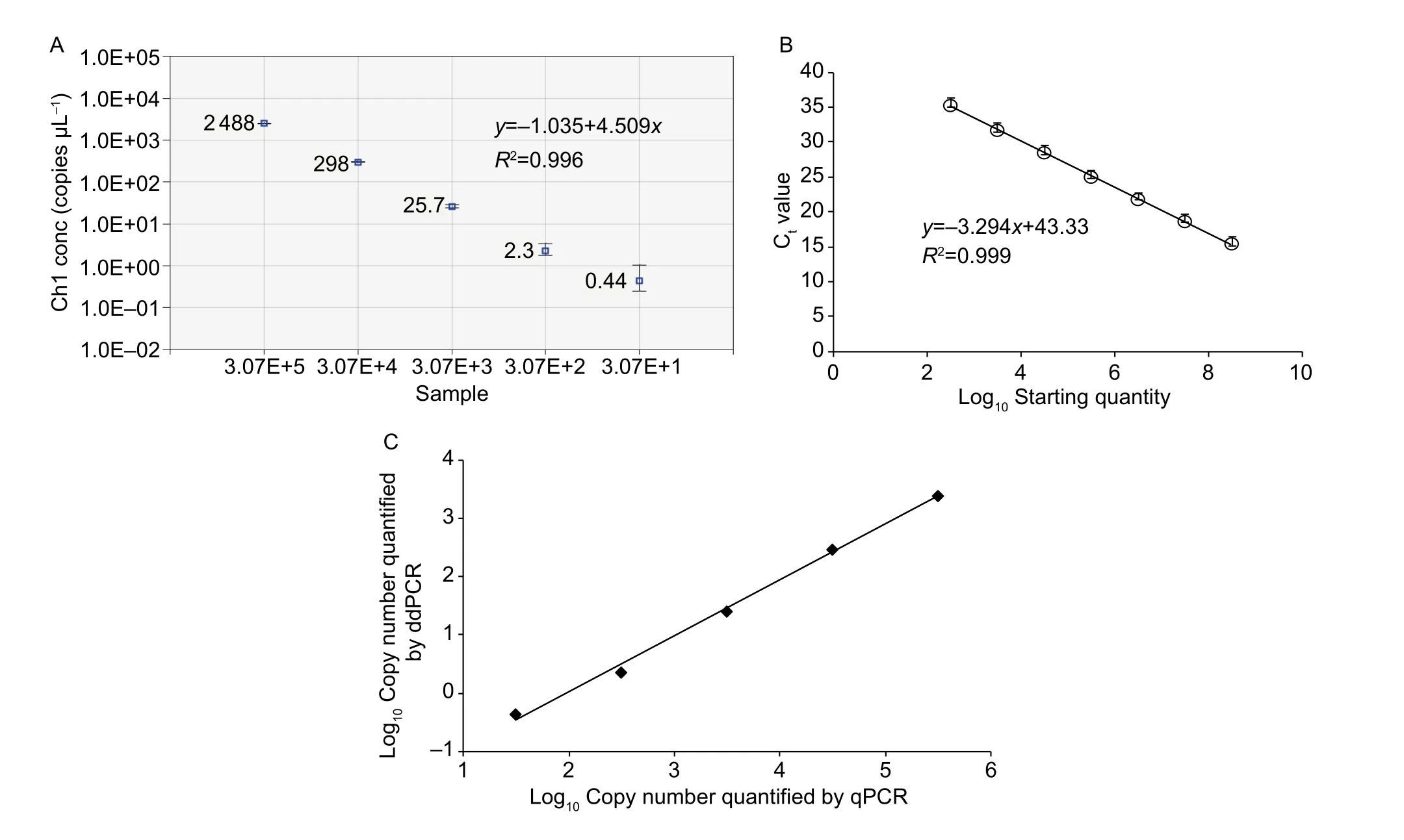
Fig. 1 Linear regression of droplet digital PCR (ddPCR, A) and real-time quantitative PCR (qPCR) assays using serial tenfold dilutions of plasmid DNA (B), and correlation between log10 means of copies using ddPCR vs. qPCR (C). Data are means±SD (A and B).
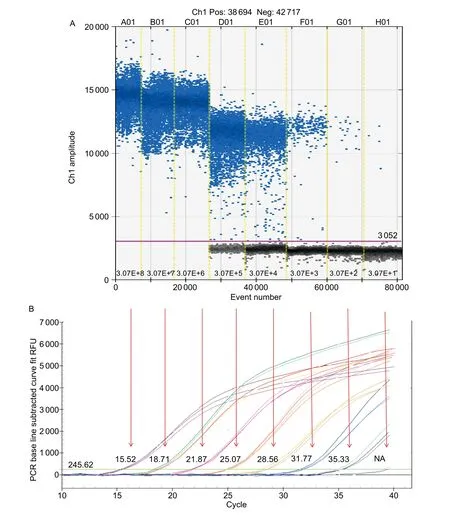
Fig. 2 Sensitivity comparison of ‘Candidatus Liberibacter asiaticus’ (Las) detection between droplet digital PCR (ddPCR, A) and real-time quantitative PCR (qPCR, B) assays. Eight ddPCR reactions with serially diluted targets are divided by the vertical dotted yellow line. The unbroken pink line is the threshold, above which are positive droplets (blue) containing the target DNA and below which are negative droplets (gray) without any target DNA. Each target concentration in ddPCR is corresponding to the Ct value(from 15.52 to NA (not applicable) in qPCR by the red arrow. RFU, relative fluorescence units.
Recently, field-capable assays, loop mediated isothermal amplification (LAMP) and serologically based immune tissue print, have been developed for Las detection (Riganoet al. 2014; Dinget al. 2016, 2017). These methods offer the advantages of simplicity, low cost and high throughput in comparison with PCR-based assays currently used.However, uneven distribution and low titer of Las in citrus plants are still big challenges for these assays. The high sensitivity ddPCR assay could be an effective complement for the detection of early HLB infection or low titer samples.
4. Conclusion
This is the first report to demonstrate the ddPCR technology for the quantification of Las. The detection sensitivity of ddPCR was compared to qPCR targeting the 16S rRNA gene. Our result showed that ddPCR was superior to qPCR for detecting and quantifying Las at low concentrations.Reducing risk of false negatives is critically important if PCR diagnosis of Las infection is used in certification programs.This methodology showed great potential for early HLB infection diagnosis.
Acknowledgements
This study was funded by the National Natural Sciences Foundation of China (31671992, 31301635), the Chongqing Science and Technology Commission Project, China(cstc2016shms-ztzx80003) and the Guangxi Key Laboratory of Citrus Biology, Guangxi Academy of Specialty Crops,China (SYS2015K004).
Bové J M. 2006. Huanglongbing: A destructive, newly-emerging,century-old disease of citrus.Journal of Plant Pathology,88, 7–37.
Ding F, Duan Y P, Yuan Q, Shao J, Hartung J S. 2016.Serological detection of “CandidatusLiberibacter asiaticus”in citrus, and identification by GeLC-MS/MS of a chaperone protein responding to cellular pathogens.Scientific Reports,6, 29272.
Ding F, Paul C, Brlansky R, Hartung J S. 2017. Immune tissue print and immune capture-PCR for diagnosis and detection ofCandidatusLiberibacter asiaticus.Scientific Reports, 7,46467.
Floren C, Wiedemann I, Brenig B, Schütz E, Beck J. 2015.Species identification and quantification in meat and meat products using droplet digital PCR (ddPCR).Food Chemistry, 173, 1054–1058.
Hindson B J, Ness K D, Masquelier D A, Belgrader P, Heredia N J, Makarewicz A J, Bright I J, Lucero M Y, Hiddessen A L, Legler T C. 2011. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number.Analytical Chemistry, 83, 8604–8610.
Jagoueix S, Bove J M, Garnier M. 1994. The phloem-limited bacterium of greening disease of citrus is a member of the α subdivision of theProteobacteria.International Journal of Systematic and Evolutionary Microbiology, 44, 379–386.
Lee J A, Halbert S E, Dawson W O, Robertson C J, Keesling J E,Singer B H. 2015. Asymptomatic spread of Huanglongbing and implications for disease control.Proceedings of the National Academy of Sciences of the United States ofAmerica, 112, 7605–7610.
Li W, Hartung J S, Levy L. 2006. Quantitative real-time PCR for detection and identification ofCandidatusLiberibacter species associated with citrus huanglongbing.Journal of Microbiological Methods, 66, 104–115.
Mock U, Hauber I, Fehse B. 2016. Digital PCR to assess geneediting frequencies (GEF-dPCR) mediated by designer nucleases.Nature Protocols, 11, 598–615.
Morgan J K, Zhou L, Li W, Shatters R G, Keremane M, Duan Y P. 2012. Improved real-time PCR detection of ‘CandidatusLiberibacter asiaticus’ from citrus and psyllid hosts by targeting the intragenic tandem-repeats of its prophage genes.Molecular and Cellular Probes, 26, 90–98.
Pinheiro L B, Coleman V A, Hindson C M, Herrmann J, Hindson B J, Bhat S, Emslie K R. 2011. Evaluation of a droplet digital polymerase chain reaction format for DNA copy number quantification.Analytical Chemistry, 84, 1003–1011.
Rigano L A, Malamud F, Orce I G, Filippone M P, Marano M R,Morais do Amaral A, Castagnaro A P, Vojnov A A. 2014.Rapid and sensitive detection ofCandidatusLiberibacter asiaticus by loop mediated isothermal amplication combined with a lateral flow dipstick.BMC Microbiology, 14, 86.
Taylor S C, Carbonneau J, Shelton D N, Boivin G. 2015.Optimization of droplet digital PCR from RNA and DNA extracts with direct comparison to RT-qPCR: Clinical implications for quantification of Oseltamivir-resistant subpopulations.Journal of Virological Methods, 224, 58–66.
Tsui N B, Kadir R A, Chan K A, Chi C, Mellars G, Tuddenham E G, Leung T Y, Lau T K, Chiu R W, Lo Y D. 2011. Noninvasive prenatal diagnosis of hemophilia by microfluidics digital PCR analysis of maternal plasma DNA.Blood, 117, 3684–3691.
Wang X, Zhou C, Deng X, Su H, Chen J. 2012. Molecular characterization of a mosaic locus in the genome of‘CandidatusLiberibacter asiaticus’.BMC Microbiology,12, 18.
Watanabe M, Kawaguchi T, Isa S I, Ando M, Tamiya A, Kubo A, Saka H, Takeo S, Adachi H, Tagawa T. 2015. Ultrasensitive detection of the pretreatment EGFR T790M mutation in non-small cell lung cancer patients with an EGFR-activating mutation using droplet digital PCR.Clinical Cancer Research, 21, 3552–3560.
Zhao Y, Xia Q, Yin Y, Wang Z. 2016. Comparison of droplet digital PCR and quantitative PCR assays for quantitative detection ofXanthomonascitriSubsp.citri.PLoS ONE,11, e0159004.
Zheng Z, Xu M, Bao M, Wu F, Chen J, Deng X. 2016.Unusual five copies and dual forms of nrdB in “CandidatusLiberibacter asiaticus”: Biological implications and PCR detection application.Scientific Reports, 6, doi: 10.1038/srep39020
 Journal of Integrative Agriculture2018年2期
Journal of Integrative Agriculture2018年2期
- Journal of Integrative Agriculture的其它文章
- Rapid mapping of candidate genes for cold tolerance in Oryza rufipogon Griff. by QTL-seq of seedlings
- A dCAPS marker developed from a stress associated protein gene TaSAP7-B governing grain size and plant height in wheat
- A major quantitative trait locus controlling phosphorus utilization efficiency under different phytate-P conditions at vegetative stage in barley
- Overexpression of IbSnRK1 enhances nitrogen uptake and carbon assimilation in transgenic sweetpotato
- Collision detection of virtual plant based on bounding volume hierarchy: A case study on virtual wheat
- lntegrated management strategy for improving the grain yield and nitrogen-use efficiency of winter wheat