
基于文献挖掘视角的组学研究脉络梳理
2018-03-21 05:09,,,,
中华医学图书情报杂志 2018年3期
,,, ,
随着科学研究的进展,人们发现单纯研究某一方向无法解释全部生物医学问题,科学家便提出从整体出发研究人类组织细胞结构、基因、蛋白及其分子间相互的作用,通过整体分析反映人体组织器官功能和代谢的状态,因此便产生了“组学”的概念。从分子生物学角度,组学主要涵盖基因组学、蛋白组学、代谢组学、转录组学、脂类组学、免疫组学、糖组学和 RNA组学等。Omics是组学的英文称谓,其词根“-ome”在英文中是指一些种类个体的系统集合。Genomics(基因组学)是最早提出的组学类型,由美国科学家Thomas Roderick于1986年提出[1],之后其他类型的组学相继出现。笔者通过查阅分析国内外大量组学相关综述后发现,现阶段的组学研究综述都是关注某一种组学的最新进展,缺少从宏观角度分析多种组学的融合研究。就目前组学研究的态势而言,多种组学技术融合已成为必然趋势。因此,全面研究组学的整体发展趋势和各类组学之间的脉络关系,显得十分重要。文本挖掘技术和信息计量学方法的发展为从海量的科研文献中梳理组学研究脉络提供了可能[2]。
文献是科研成果的主要产出和表达形式,是由科研工作者对其创造性研究成果进行理论分析和科学总结并公开发表的文体,也是医学事业不断发展的重要科技信息源,是记录医学科技进步、重大发明和改革的历史性文件[3]。文献挖掘[4]是数据挖掘领域中一个重要研究方向,其处理对象是文本类型的文献数据。一般通过统计方法获取所关注的文献,再使用自然语言处理方法从中抽取出特定的事实信息,并对内容进行分析,从非结构化的数据中分析出隐藏的一些规律。文献挖掘方法已在多个领域中得到了广泛的应用,如生物学、医药学、生物医药学以及科学计量学等。文献挖掘技术[5]主要包括信息检索、实体识别和信息抽取。实体识别[6]旨在发现文献中重要的实体,该技术中常见的方法为基于特征、基于词典或者基于规则进行实体识别。而信息抽取技术主要把文献中含有的重要信息或者事实抽取出来,并用形式化的结构表示,依据共现关系[7]和自然语言处理技术[8]进行文本内容关系的抽取。
文献计量分析[9]有助于全面了解某一研究领域的国内外文献发表情况,目前以所有组学为对象的文献计量分析少之又少。通过分析国内外文献发表情况,方便该领域研究人员了解组学的研究现状及发展方向,有助于科研管理机构在项目评审、资助中合理分配资源,有助于其科研选题、成果发表及选择研究合作方并调整研究方向[10]。
本文拟利用文献计量学方法,借助PubMed数据库及相关文献挖掘、分析方法对“组学(Omics)”相关英文文献进行统计和分析,探寻组学的研究轨迹,为研究人员更加深入系统地开展组学研究提供参考。
1 资料来源与方法
本文使用的数据集来自PubMed数据库[11]。美国国立生物技术信息中心(National Center for Biotechnology Information,NCBI)提供的The Entrez Programming Utilities(E-utilities)编程工具,是访问NCBI Entrez查询和PubMed数据库的稳定接口,可以实现PubMed数据库记录的批量下载。本文使用E-utilities中的Esearch和Efetch 2种工具获取PubMed记录,时间跨度为1896-2016年,共获得27 040 819条记录,包含所有的出版类型。本文关注的主题为“组学(Omics)”。组学是研究一些种类个体的系统集合的学科,如基因组是构成生物体所有基因的组合,基因组学这门学科是研究这些基因以及这些基因间的关系,因此我们将组(Omes)与组学(Omics)同等对待。具有组学含义的单词均有一个共同的特征,即以“-ome” “-omes”“-omic”或“-omics”结尾,故本文选取文献的“title”或“abstract”为统计窗口,从中识别具备上述特征的单词,在数据集中共识别出19 268个具备上述特征的单词。通过删除噪音单词(如“some” “home”等),最后得到77个出现频次不低于10次的“-Omics”单词(表1),以供下一步分析。将上述77个“-Omics”单词重新在原始数据的题目和摘要中用Python语言编写的程序进行匹配,27 040 819条原始数据中含有77个“-Omics”单词中的任何一个的记为有效数据,共得到346 977条记录作为本文的数据集。
可视化分析采用VOSviewer,它是一款用来构建和查看文献计量图谱的免费文献计量分析软件,基于文献的共引和共被引原理,可用于绘制各个知识领域的科学图谱。将所有类型组学的共现数据经过处理后导入VOSviewer进行可视化,得到网络可视化图。图中圆圈和标签代表关键词,圆圈及标签大小代表其重要性的高低,拥有相同颜色的圆圈属于同一个聚类[12]。
主题河是一种被证明为可有效反映文本之间的时间属性的方法。在这种可视化方法中,时间被表示为从左往右的一条水平轴,然后用不同的颜色条带代表不同的主题,条带的宽度代表该主题在该时间的一个度量。这样人们可以跟踪任何一个主题在量上随时间的变化,也能比较不同的主题在同一个时刻相对规模的大小[13]。
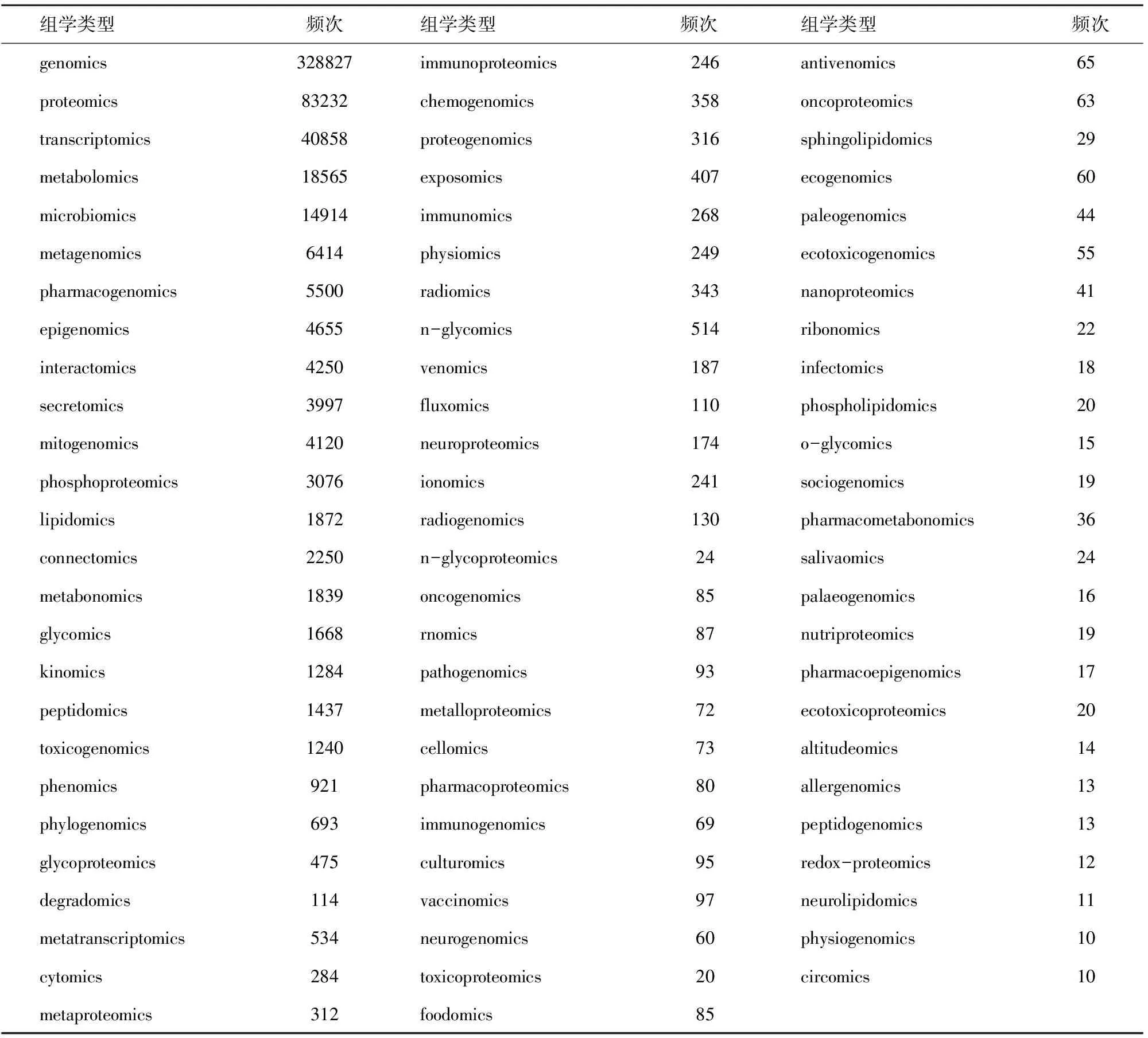
表1 出现频次不低于10次的“-Omics”单词
2 结果与分析
从上述数据集中筛选出关于组学的相关文献共计346 977篇,包括期刊论文345 549篇(占99.59%)和综述1 428篇(占0.41%)。
2.1 文献年度变化趋势
文献的年度分布情况可以从一定程度上反映该领域的发展情况。分析文献量与时间变化的关系可以反映研究主题的发展情况,可以大体揭示该主题的发展阶段与规律。本文将组(-ome/-omes)与组学(-omic/-omics)同等对待,美国科学家Thomas Roderick于1986年最先提出的是Genomics(基因组学),而第一篇提到“基因组(genome)”的文献则出现在1943年。1943-2016年全世界组学相关文献发表情况如图1所示。从1943年之后组学相关研究的发表量整体呈逐年递增趋势,从1999年的4 331篇迅速增长到2000年的5 288篇,到2016年文献发表量已达40 590篇。
人类基因组计划(Human Genome Project,HGP)由美国于1987年启动, 2000年6月26日参加人类基因组工程项目的美国、英国、法国、德国、日本和中国等6国科学家共同宣布,人类基因组草图的绘制工作已经完成,后基因组时代来临。组学领域的研究文献呈现了井喷式的增长,已有越来越多的国内外科学工作者投入到组学研究中,并获得了大量的研究成果,组学已逐渐成为生物医学研究领域的热点之一。
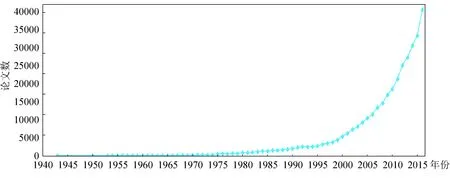
图1 组学研究论文发表情况
2.2 各类组学文献情况
2000年之前组学研究类型较单一,之后各类组学研究相继涌现,并呈现出不同的变化。我们选取数据中论文总数排名前10的组学类型进行比较,发现各类组学都呈现了逐年递增的现象。其中基因组学的文献发表量遥遥领先,蛋白质组学和转录组学的文献发表量紧随其后。2000-2014年,蛋白质组学的文献发表量一直高于转录组学,2014年之后转录组学的文献发表量赶超了蛋白质组学。原因在于,从2008年开始,第二代测序技术利用一系列高通量测序技术(high throughput sequencing)进行大规模的基因组DNA或RNA测序,能够快速准确地获得基因组编码序列,满足极短时间内对基因组进行高分辨率检测的要求。随着第二代测序技术高通量、高准确率、低成本等优点的实现,转录组学测序技术也随之得到了更广泛的应用[14]。因此,转录组学的关注度逐渐升高并且超过了蛋白质组学的关注度。
2.3 各类组学共现情况
统计不同类型的“组学”之间在同一篇文献的题目和摘要中出现的情况,便可形成多组学研究的相关关系。将多组学共现类型细分为在同一篇文献中分别出现2种类型、3种类型、4种及4种以上类型,并进行分类计量。1995年首次出现多组学共现的文献。2种类型组学共现的文献量一直处于遥遥领先的状态,3种类型组学共现和4种及4种以上组学类型共现的文献量较2种类型组学共现的文献量还有一些差距,但总体来说各种共现情况都随着时间的增长呈现出逐年递增的趋势。
将多组学共现数据导入VOSviewer中,其结果以可视化图谱的形式展示出来。如图2所示,可以看出基因组学、转录组学、蛋白质组学、代谢组学是组学共现研究的热点,文献数量居于前列。通过连线可以看出,基因组学与转录组学的共现文献量最多,基因组学与蛋白质组学的共现文献量次之。各类组学之间都存在着错综复杂的关系。
研究结果表明,多组学的结合研究已成为组学研究领域的趋势,整合多组学数据用于药物重定位和个性化医疗越来越受到重视[15]。因此,相关领域科研人员未来要注意多组学类型的结合研究,从而促进组学研究的进一步发展。
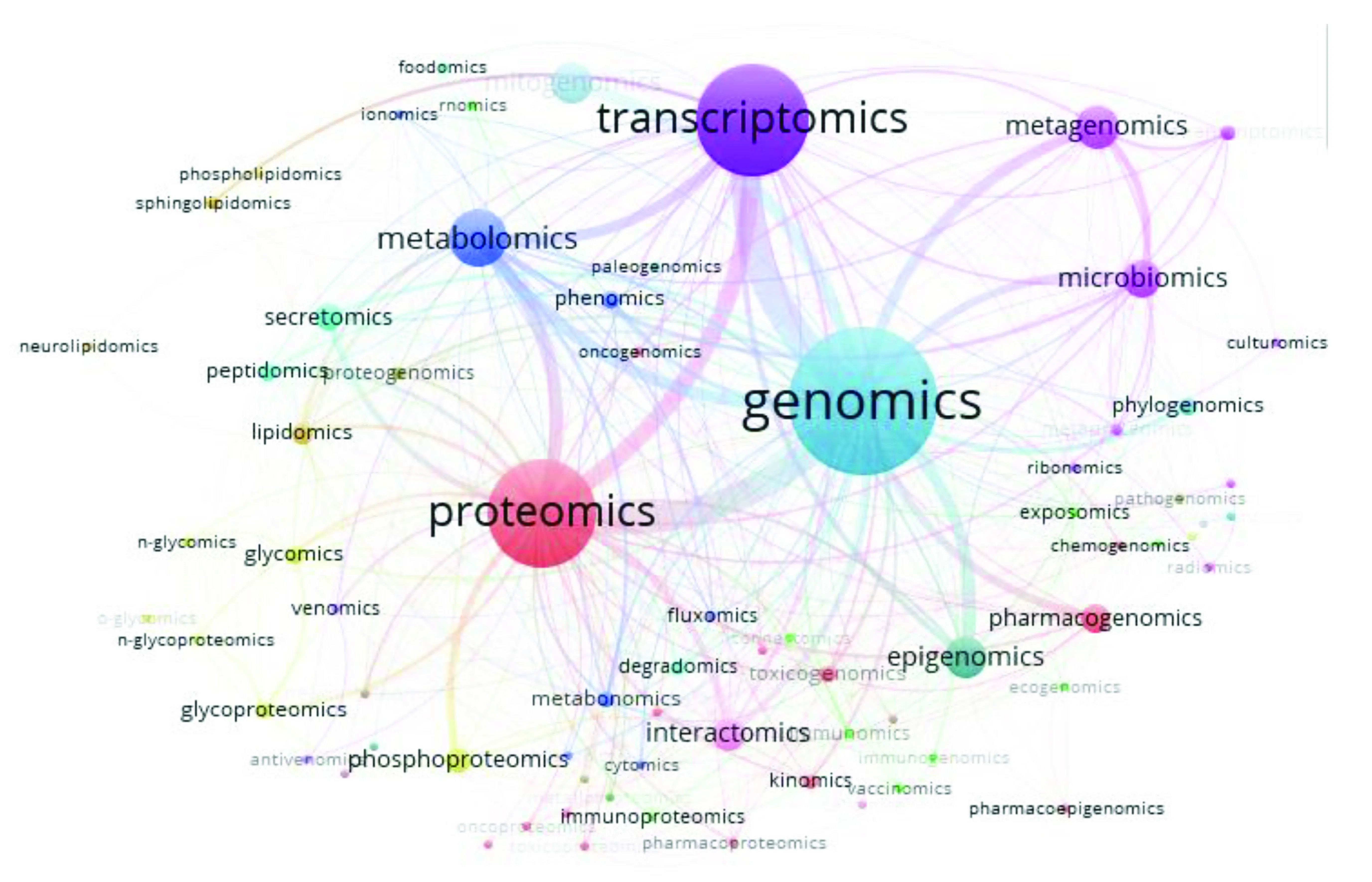
图2 所有类型组学共现情况
2.4 基因组学脉络研究
“基因组学”为最早出现的组学类型,且与各类组学都有共现的情况,因此以“基因组学”为主脉络,展示其余各类组学与“基因组学”共现研究的相关情况,通过主题河图进行呈现。选取与“基因组学”共现文献总量排名前15的组学类型,年份从出现多组学共现的第一篇文献的1995年到2016年进行研究(图3)。图3中河流的宽窄代表各类组学与基因组学共现的文献数的比例,横坐标为年份的变化。

图3 “基因组学”与其他各类组学共现论文数变化情况
2.4.1 稳定型增长类型
最早与“基因组学”共现的是“蛋白质组学”。“蛋白质组学”这个概念由Marc Wikins 1994年首次提出[16]。在1995年“基因组学”与其他类型组学共现的5篇论文中,4篇是“基因组学”与“蛋白质组学”的共现,“蛋白质组学”与“基因组学”的共现论文数一直处于稳定增长的趋势。究其原因,一方面,从分层递阶结构来说,蛋白质系统的粒度较基因组系统粒度粗,蛋白质系统数据处理的复杂度不会超过基因组系统数据处理的复杂度;另一方面,蛋白质的功能性研究距离我们所期望的在细胞水平上研究分子生物学更近,或者说距离在实际应用中所需要的功能研究更近,如在药物基因组学中的关键蛋白质组的寻找[17]。
2.4.2 井喷型增长类型
“转录组学”与“基因组学”的共现文献量随时间的变化呈井喷式增长,到2016年已成为与“基因组学”共现占比最大的基因类型。究其原因,一是转录组学是功能基因组学研究的重要组成部分,是一门在整体水平上研究细胞中所有基因转录及转录调控规律的学科[18-19];二是随着新一代高通量基因测序技术运用到转录组学研究之中,转录组学研究中提供的数据量呈现爆炸式的扩增,拓宽了转录组学研究解决科学问题的范围[14]。
“线粒体基因组学”与“基因组学”的共现论文数量从1995年到2014年一直处于缓慢增长态势,然而到2015年共现文献量呈现井喷式增长,成为2016年当年排在“转录组学”之后的第二大共现组学类型。究其原因,是由于“线粒体基因组学”在2008年后随着中国科研人员的加入,半翅目昆虫线粒体基因组测序进入了迸发阶段,在2008-2015年共获得了89种昆虫的线粒体基因组,其中81种在中国完成测序。截至2015年5月,美国国立生物技术信息中心共收录100种半翅目昆虫的线粒体基因组,其中83个为全线粒体基因组,17个近似完整的线粒体基因组[20]。线粒体基因组的获取完成在极大程度上推进了线粒体基因组学与基因组学的共同研究。
3 结束语
文献资料中涵盖了大量重要信息,能够从海量的文献资料中快速挖掘出人们所需求的信息知识,是文献挖掘技术日益受重视的主要原因。我国“文献挖掘”多采取在数据库中检索所研究的主题对结果进行分析的方式。本文采用获取PubMed数据库1896-2016年的全数据的方法,通过对所研究主题的词根进行识别挖掘,运用社会网络分析的方法和可视化技术,从组学相关文献的年度变化趋势和共现情况方面进行分析,为传统的文献挖掘提供了一种新的思路,为学者和研究人员创造了一个知识共享平台。同时通过分析研究数据,发现后基因组时代的到来把组学研究推向了高潮,无论是数量还是种类都出现了井喷式的增长。多类型组学的融合研究越来越受科研人员的关注,已成为未来组学研究的热点趋势。本文的不足在于只从英文文献着手,研究方法还不够完备,对多种类型的数据的处理与挖掘还不完善。
猜你喜欢
昆明医科大学学报(2022年3期)2022-04-19
今日农业(2021年11期)2021-08-13
昆明医科大学学报(2021年4期)2021-07-23
智慧健康(2021年33期)2021-03-16
今日农业(2020年19期)2020-12-14
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
科学(2020年2期)2020-08-24
科学(2020年2期)2020-08-24
中成药(2018年7期)2018-08-04
