
质谱图聚类网络法在鉴定多肽翻译后修饰中的应用及研究进展
2018-10-25 09:11何明敏舒坤贤白明泽许睿
生物工程学报 2018年10期
何明敏,舒坤贤,白明泽,许睿
重庆邮电大学 生物信息学院 大数据生物智能重庆市重点实验室,重庆 400065
蛋白质组学是研究生物系统中的所有蛋白质的科学,肽组学是研究生物体内源性低分子量蛋白质或多肽及其变化规律的科学。基于蛋白质组学和多肽组学的质谱分析技术在研究蛋白质或者多肽中逐渐成熟。现代质谱仪的飞速发展使得每天能产生数以百万计量的串联质谱数据,再加上软件算法和计算能力的发展,高通量蛋白质组学研究得以实现。实际上,存储注释或者未注释的质谱以及相应多肽及其翻译后修饰(Post-translational modifications,PTMs)信息的数据量在成倍增长。比如蛋白质组学相关数据库GPMDB[1]、Uniport(http://uniprot.org)、PRIDE[2]以及一些特殊内源性多肽数据库 Neuropeptide(http://neuropeptides.nl/)和SwePep(www.swepep.org)[3]等。
在蛋白质组学的鉴定中,一个比较大的挑战是多肽的翻译后修饰鉴定[4]。PTMs广泛存在于真核细胞生物中,对生物体的信号传导以及生命活动至关重要,但是 PTMs鉴定往往比未修饰多肽鉴定更加困难。
本文首先介绍了质谱网络的发展历程,以及基于谱图聚类思想的质谱网络的建立,然后详细讨论了利用质谱网络如何进行非预设的翻译后修饰鉴定;最后结合质谱网络方法鉴定内源肽的实际应用,讨论了质谱网络方法所面临的挑战和发展方向。
1 质谱网络方法的发展
1.1 蛋白质组学中的多肽鉴定方法
现有的质谱鉴定方法主要采用3种方式来实现,分别是传统的序列数据库搜索法、谱图库搜索法和从头测序法(de novosequencing)。
使用传统的序列搜库方法成功鉴定出的质谱和相关肽段的数据量在飞速增长,这种主流方法是已使用近20年的经典方法:采用胰蛋白酶消化所提取的蛋白质得到肽段,然后通过串联质谱法产生肽的串联质谱[10],接着将谱图与蛋白质序列数据库进行匹配计算,以判断该质谱是否由某肽段所产生,最终获得肽和蛋白质的鉴定。虽然这个过程看起来很简单,容易实现,但在实验中鉴定蛋白质或者多肽时,因为化学噪声、修饰、肽段离子的不完全破碎、污染等问题,使得鉴定过程仍然是一项非常复杂的任务。此外,因为蛋白质数据库的不完整性,可能不存在对应于实验谱的理论谱数据,这些研究困境导致很难产生满意的研究结果。
目前主流的开源蛋白质序列数据库搜索工具见表1。用于序列数据库搜索的软件工具除了表1中提到的,还包括非开源搜索引擎SEQUEST[11]、MASCOT[12]、P-Mod[13]、Interrogator[14]、TwinPeaks[15]、SeMoP[16]和 PTMap[17]等。

表1 开源蛋白质序列数据库搜索工具一览表Table 1 Protein sequence search tool list
质谱谱图库搜索方法是另一种多肽鉴定方法,其鉴定流程如图1所示。从谱图库中提取高质量鉴定结果,利用谱图搜索工具建立一致性谱图库,从谱图库中选择一致性谱图作为候选谱图,然后将实验获得的质谱谱图与候选谱图进行匹配以完成对实验谱的鉴定。这种谱图库搜索方法的基本理论假设是特定有机分子在质谱仪中会以稳定的方式碎裂,并且同一分子会有相同或相似的碎裂形式,即能得到相似的谱图。谱图库搜索方法实际上是实验谱与实验谱之间的匹配。谱图库搜索可以充分利用所有质谱特征,包括实际峰值强度、片段的中性损失以及各种不常见甚至未表征的片段,根据这些特征来确定最佳的匹配。提升图谱比对相似性打分算法的精确性,且将搜库空间限制在已鉴定肽段产生的谱图,极大地缩小了搜索空间,使得选库更加灵活,搜库更有选择性,从而显著减少耗时。最后,通过已有的鉴定结果构建一致性图谱库,不需要额外的时间和功耗就可以整合不同方法鉴定的肽段信息。与序列搜索相比,谱图库搜索方法大大缩小了搜索空间。目前主流的存储实验质谱的谱图库如表 2所示。

图1 谱图库搜索流程Fig.1 Spectra library searching process.
这两种搜库方法各有优点。在谱图库搜索中,将先前实验中观察和鉴定的肽段存储在谱图库中并认定其为候选物,而在序列库搜索中,所有推定的肽序列和翻译后修饰位点的所有信息都可能被收集在候选肽库中,而实际上因为搜索空间太大、错误率太大等各种原因,序列库搜索中考虑的这些可能出现的肽离子,其中大多数在实践中未曾被发现过。因此,理论上质谱库搜索的搜索空间可以减小几个数量级,搜索的速度相应也可以提升几个数量级。谱图库搜索方法需要有很多先验的谱图数据,如果更换实验仪器就需要使用新的先验谱图库,对实验仪器的依赖性很强。所以这种方法常用于鉴定特殊产物。如果已知的数据是已经确定的某种实验仪器产生,那么选择这种方法进行谱图库搜索鉴定未知聚类簇中的质谱是十分可行的。
目前主流的开源蛋白质谱图库搜索工具如表3所示。谱图库搜索的方式是对于已有准确鉴定结果的质谱数据作为先验数据,对待鉴定的质谱进行整体相关性比较。使用整谱比较的方法在噪音数据的过滤方面没有进行有效的处理,鉴定方法的灵活性差,鉴定准确率低且鉴定速度慢。通常,只有最多30%的MS/MS(串联质谱) 以高置信度被识别[18]。
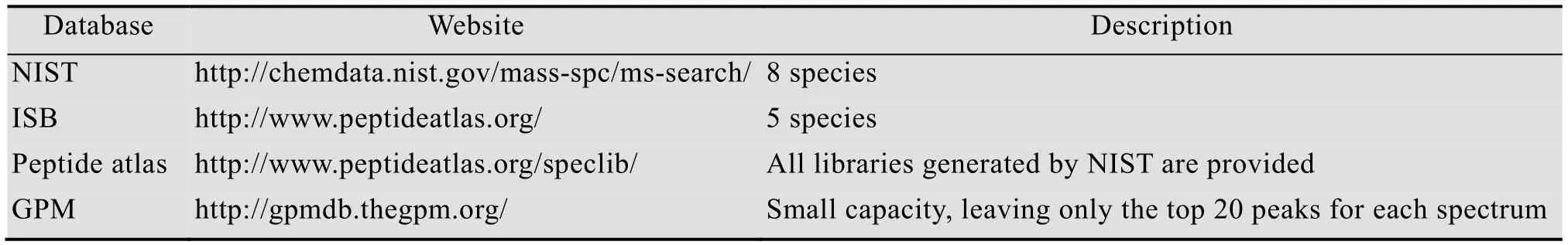
表2 蛋白质组学谱图库列表Table 2 Proteomics spectra database list
前两种方法都依赖于既有知识,第3种方法,即基于从头测序(De novosequencing)[19]则不需要。这种方法被认为是在包含所有可能肽的搜索空间进行肽搜索。从头测序法将数据库中的含有可能修饰的氨基酸序列构建序列标签,然后根据全肽序列与观察到的序列质谱谱图之间的剩余质量差来推断潜在的修饰。从头测序常被用于研究来自未知蛋白质的质谱图。由于测序准确性的困难,完全自动化的从头测序分析仍然是一个难以实现的目标,单个离子阱串联质谱的传统算法在预测鉴定时,鉴定结果中每4个氨基酸中仍然可能存在一个不正确的氨基酸(即公布的从头测序算法的准确率只有75%)[20]。使用目前已有的高分辨质谱进行从头测序,准确率提高到了84%[21],使用机器学习方法的准确率可以达到88%[22]。经典的基于鸟枪法的蛋白质从头测序的方法,从串联质谱组装成氨基酸序列时需要经历3个步骤:1) 使用质谱图比对寻找到来自重叠肽的质谱图对;2) 组装比对对齐的谱图;3) 确定每组组合谱图的一致性氨基酸序列。基于从头测序方法的软 件 有 Lutefisk[23]、 PEAKS[24]、 PepNovo[25]、UStag[26]、MODa[27]和 pNovo[28]等。
质谱网络方法属于谱图库方法的延伸。该方法不仅能够解释来自重叠多肽之间的谱图比对(Aligned),还能对有修饰多肽和无修饰多肽之间的谱图进行比对。在鸟枪法蛋白质测序中,质谱网络实现了有史以来报道的离子阱数据中的最长序列和准确率最高的从头测序序列[29],另外,质谱网络将相同多肽的多个修饰变体和未修饰变体的谱图组合分析,直接从实验数据中发现修饰和高度修饰的多肽。实现该算法的开源代码可以从peptide.ucsd.edu 下载[30]。
1.2 质谱网络方法的发展
质谱网络的发展可追溯到质谱库的发展,最早关于谱图库搜索的研究思想在1988年被Yates等提出[34]。利用质谱图库可以进行待鉴定实验质谱与质谱的匹配比较,从而帮助实现多肽的鉴定。
在鉴定多肽时,其中很重要的 PTMs的鉴定会明显增加鉴定的难度。如前所述,基于质谱的蛋白质/多肽鉴定主要有3种方式,这3种方式中鉴定PTMs的方法各有不同。
在数据库搜索中,将每个串联质谱已知多肽序列的给定数据库进行比较,并选择显著的匹配用于蛋白质鉴定。对于 PTMs的鉴定,如果不限定修饰类型,在鉴定多肽时将大大降低搜索速度和鉴定准确度。因此,使用该方法时建议每个肽只允许一个非预设修饰,即必须事先指定修饰类型[35]。
谱图库搜索方法鉴定PTMs时,将已经被鉴定出来的质谱作为参考谱图库,以开放式搜索模式搜索鉴定非预设的 PTMs。pMatch[32]就是这样一个用于开放式谱图库搜索的工具。谱图库方法鉴定PTMs时,实际依赖库中的先验PTMs鉴定,对于库中未出现的PTMs无法发现。

表3 开源蛋白质谱图库搜索工具Table 3 Protein spectrum search tool list
多个研究小组已经公开了使用质谱图比对作为鉴定非预设的翻译后修饰的方法。该想法被Bandeira等提出的质谱网络[36]的概念很好地实现。质谱网络[37]基于重叠肽之间、有修饰和未修饰之间的质谱比对,不依赖于数据库中的先验PTMs就能鉴定出PTMs。
在非限制性翻译后修饰(Unrestrictive PTMs)鉴定方法中,质谱网络方法主要使用质谱图对来鉴定未修饰的肽。质谱网络方法基于图谱匹配策略,能够鉴定非限制翻译后修饰,不需通过搜索谱图库来获得肽段或者谱图匹配的信息,而是直接从实验谱图中搜索修饰肽段与非修饰肽段的图谱对以获得修饰信息[38]。
1.3 质谱网络库的建立
质谱网络方法鉴定蛋白质/多肽的第一步是建立质谱网络库。质谱网络库的建立流程如图 2所示,实验获得的串联质谱数据通过谱图质荷比、峰的强度之间的相似性进行聚类[39],获得不同的簇,由簇与簇的一致性谱图之间的关联性建立质谱网络,最后整理所有质谱网络,整合数据集,利用数据库搜索方法建立质谱网络库。
1.3.1 质谱网络的建立
酶切后的蛋白质样品通常含有多个重叠的多肽。建立质谱网络的第一步是建立质谱图对,然后从多个质谱图对中找寻质谱星,最后利用各个质谱星和质谱图对构建质谱网络。
首先定义肽对,肽对的定义有两种方式,一是相同肽不同的修饰或者突变,二是肽P1是肽P2的前缀或后缀,则肽 P1和肽 P2组成肽对[40]。如果两个质谱图对应的肽配对,则可以看作这两张谱图能够形成质谱图对。质谱图对通常来源于重叠的肽或者同一条肽修饰和未修饰的变体。
质谱图对的产生,打开了一种新的计算途径。一对质谱图对允许分离b(前缀质量) 和y(后缀质量) 离子质量梯,大大地减少噪声峰的数量,以及将修饰的鉴定从已鉴定谱图传播到未鉴定谱图,从而将非预设的PTMs检测出来。
入射到质谱图对中谱图S1的一组谱图称为质谱星(Spectral star)。即使对于单个质谱图对(S1,S2),质谱S1和S2的b离子(y离子) 已经具有高的信噪比和丰富的前缀和后缀。质谱星允许进一步丰富谱图的前缀和后缀。由质谱图对(S1,S2)、(S1,S3)……(S1,Sn)组成的质谱星通过考虑S1和S2产生的 2(n-1) 个 b离子和 y离子的比较(2≤i≤n))来增加信噪比。使用聚类方法将所有这些谱图组合成质谱星S*。从质谱图对(Si,Sj) 和质谱星(Si*) 得到的高质量的谱图使得这些谱图的解释更加简单明确。 由于这些谱图具有前缀梯度和后缀梯度以及极易分离的少量噪声峰值,所以这些谱图的从头重建(De-novo) 产生的正确标签包含十分可靠的长序列。平均来说,一致性谱图的从头测序能正确地识别长度为n的肽中 72%的结果,这已经是非常高的识别率,因为第一个(例如,b1) 和最后一个(例如,bn-1) b离子很少存在于 MS/MS质谱图中,所以几乎不可能在样品中解释超过80%的切割肽。在实验中,除了最佳的从头重建之外,还能够产生次优重建以及长肽的标记物[25]。
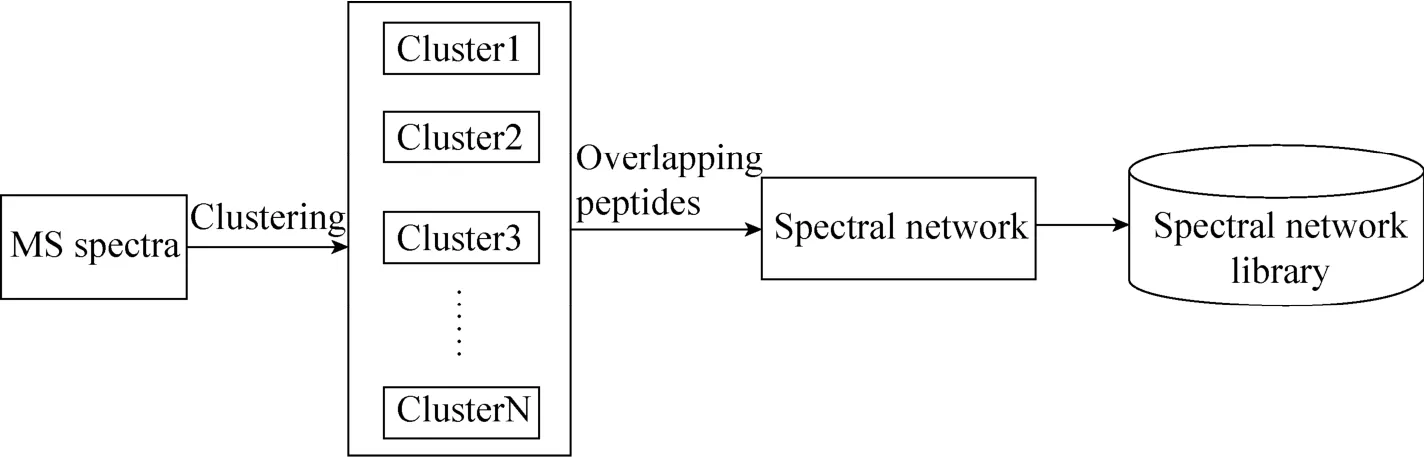
图2 质谱网络库建立流程Fig.2 Process of establishing the spectral network library.
使用特异性酶进行酶切的蛋白质样品通常含有覆盖蛋白质序列相同区域的多个重叠肽,例如前缀肽(Prefix peptides)(例如PEPTI/PEPTIDES)、后缀肽(Suffix peptides)(例如TIDES/PEPTIDES)或部分重叠的肽(例如 PEPTIDES/TIDESHIGH)。如果肽序列是预先已知的,可直接应用标准序列比对算法确定它们的重叠部分。类似地,质谱图比对被定义为来自重叠肽的谱图之间的对应峰的对准。相较于序列比对,在质谱网络中,质谱图比对不用预先知道肽序列,可充分利用重叠肽部分氨基酸的重合,在b 离子和y离子的质量中编码的序列信息足以检测成对的来自重叠肽的串联质谱。事实上,质谱图比对具有较高的可靠性,能够在高通量蛋白质组学实验中从数百万可能的质谱图对中辨别高分值的真实质谱图对。此外,由于每张谱图可以与几个其他谱图比对,所以检测到的质谱图对的集合定义了质谱网络:其中每个节点对应于不同的质谱图,如果发现相应的质谱图被显著地比对上,则通过边缘连接节点。值得注意的是,由于大多数质谱图通常来自非邻接蛋白质区域,这种方法产生的不是单个质谱网络,而是来自重叠肽的每组质谱图形成的多个质谱网络。
首先,质谱网络基于质谱图比对而不是与蛋白质序列匹配。第二,质谱网络在考虑其可能的鉴定之前,能找到来自相关肽的质谱图。第三,质谱网络可从相关肽的质谱集合确定一致性标识质谱,而不用每次分别尝试识别一个质谱图。质谱网络库允许在没有任何数据引用的情况下检测修饰,即不依赖于数据库。这是与谱图库搜索方法最大的区别。此外,与标签技术[41]相比,质谱网络方法不是以受控制的方式引入修饰,而是利用样本中自然存在的多种修饰用于解码未知的修饰。质谱网络方法弥补了数据库搜索方法的限制,它在赋予肽的修饰注释信息时更具有选择性。
1.3.2 质谱网络库的优势
前文提到质谱网络由来自重叠肽的匹配谱图构建。Bandeira等发现仅由同一条多肽产生的不同的翻译后修饰质谱图对就能表现出相似的碎片离子化模式[40]。使用质谱网络分析串联质谱图主要有 3个方面与主流数据库搜索方法不同。
质谱网络将来自相同肽的修饰或者未修饰的多个变体(Variants) 的谱图分组在一起,质谱网络有助于可靠地鉴定高度修饰的多肽。尽管数据库搜索仅限于理论谱图和实验谱图之间的离子质量匹配,但质谱网络进一步利用了离子在相应质量和相似峰强度下的相关信息。一般而言,如果另外观察到与中间修饰状态的肽产生的谱图高度相似,则更容易鉴定出高度修饰(多重修饰) 的肽。因此,质谱图比对不仅可以发现非预设的修饰,而且还为鉴定高度修饰的肽提供了参考方法。
1.4 翻译后修饰发现的鉴定
目前,重要的体内修饰研究主要有磷酸化、甲基化、糖基化、泛素化等。当首次分析可能含有修饰肽的样品时,人们并不知道哪些残基或肽将被修饰。数据库搜索鉴定多肽时,如果不限定修饰类型,则需要考虑所有可能的位点之间的质量差异(修饰质量)。
质谱网络中通过质谱图比对发现PTMs的基本思想参考[37]图3所示,对多肽的修饰表现为谱图峰的质量差,当把这个质量差考虑进质谱峰的匹配时,两张谱图可以很好地比对上。这种方法要求比对的谱峰之间显著匹配[37],但不限制要考虑的修饰类型,可以用来发现全新的或非预设的修饰。
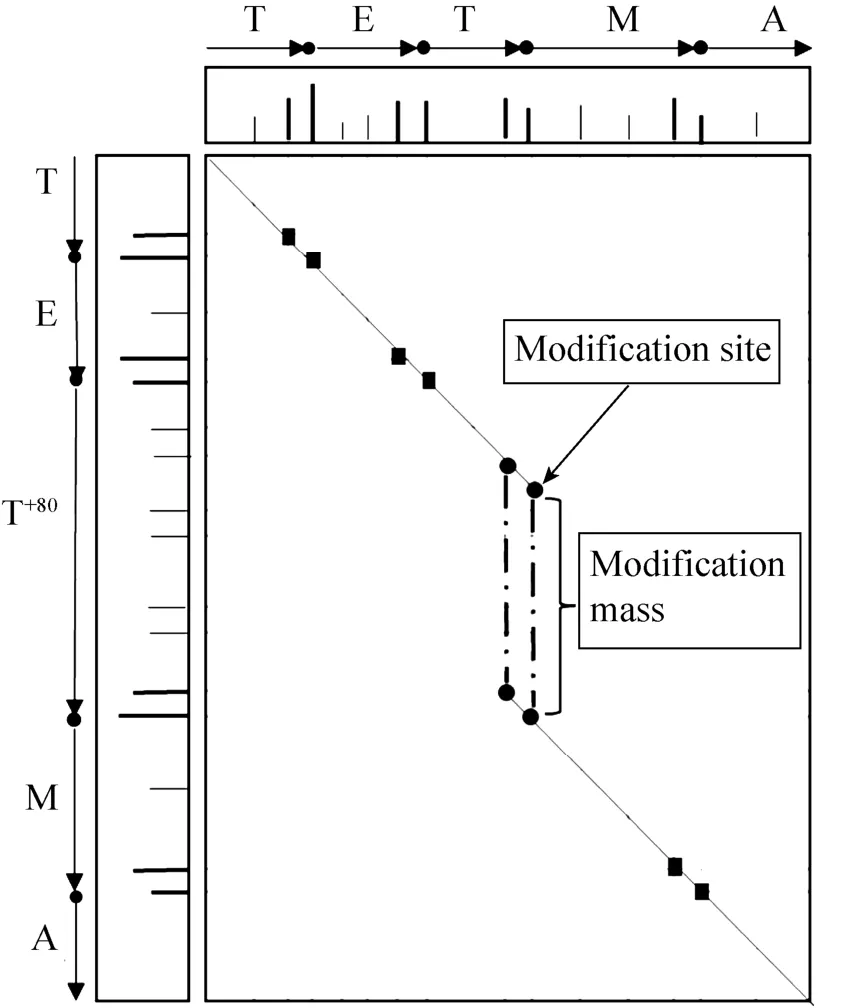
图3 肽TETMA的修饰和未修饰变体之间的质谱图比对Fig.3 Spectral alignment between modified and unmodified variants of the peptide TETMA.
非限制性翻译后修饰搜索不事先指定修饰类型,可快速发现潜在的修饰类型和修饰位点,或者发现新的修饰类型(非预设的修饰发现)。质谱网络方法中,利用样本中发生在肽自身的修饰,不需要事先指定修饰类型。并且可用于检测仅发生在少量肽上的修饰,这些修饰往往不太可能被PTMs矩阵检测方法检测到[42]。
使用谱图库搜索方法鉴定非限制性PTMs常用的软件有:Inspect[43]、pMatch[44]、SpectraST;而使用传统的序列数据库搜索方法鉴定限制性PTMs时所用的搜索引擎有:SEQUEST[11]、pFind[45]、Mascot、PEAKS[46]、X!Tandem、MaxQuant[47],其中几项主流软件 Comet、Mascot、Sequest、SpectraST、Tandem 整合在 TPP(Trans Proteomic Pipeline)[48]中。
利用质谱网络可用于检测非预设的翻译后修饰,搜索中不需要事先指定修饰类型,即可快速发现预料之外的修饰类型和修饰位点,甚至可能发现新的修饰类型。InsPect[43]为常用的非限制性翻译后修饰搜索引擎。
使用数据库搜索鉴定含有多重修饰的肽时,如果所有肽的可能修饰的数量组合出现,将是一个巨大的计算挑战问题。计算速度慢仅是一方面的问题,随着肽数量的增加,给定质谱图的假阳性识别的风险也快速增加。然而,含有两个或多个修饰的肽通常还含有仅具有一个或没有修饰的相同肽的变体,在这种情况下,质谱图比对能将来自相同肽的多个修饰变体的相关质谱分组为小质谱网络,从而增加它们的置信度。
通过将来自相同肽的多个变体的质谱分组在一起,说明质谱网络有助于修饰肽的可靠鉴定。虽然数据库搜索方法局限于理论和观测质谱之间的匹配离子质量,但质谱网络进一步利用了相似峰强度对应质量的共同碎片离子来鉴定。如果从中间修饰状态能够观测到高度相似的质谱,则更容易鉴定高度修饰的肽。因此,质谱图比对不仅可以发现非预设的信息,而且还提供了用于鉴定高置信度修饰的肽的替代途径。
2 质谱网络方法所面临的挑战
质谱网络算法之前主要被应用于单个实验数据集的鉴定中,如使用IKK β(IKK蛋白β亚基)数据集[43],建立质谱图对,对重叠肽的鉴定[49],也曾成功鉴定出之前未曾鉴定到的 PTMs。但是迄今为止质谱网络还未被应用到大规模的质谱库里进行“大数据”的挖掘应用。PRIDE Cluster是基于MS-Cluster算法,对PRIDE Archive保存的大量质谱数据进行聚类分析后建立的质谱归档库[50],它成功将正确鉴定之外的质谱数据分成了3类:a) 错误鉴定的质谱;b) 正确被鉴定但是却低于阈值的质谱;c) 未鉴定的质谱数据。对占据总量一半以上的c类数据,研究基于聚类时所获得的相似性数据成功鉴定出了160条肽段,但是大部分仍是酶切片段。我们推测,在c类数据中还有大量的非酶切肽段等待质谱网络方法来鉴定。
质谱网络库是基于谱图库概念基础上建立起来的,它不仅存储已鉴定的质谱(即质谱图库),还存储未鉴定的质谱,以及保留了关于在各物种和各条件下常见的肽段质谱的信息。因此,质谱库不仅提供了传统的基于谱图库和质谱相似性的搜索能力,还提供了对分析数据的新方法的支持。
用质谱网络方法来分析串联质谱数据与传统分析方法有3个地方不同:1) 质谱网络将已鉴定的实验质谱与其他未鉴定的实验质谱相匹配,而不是与蛋白质序列产生的理论质谱进行匹配;2)在考虑质谱可能获得的鉴定之前,质谱网络就能发现它与相关肽段的关系;3) 质谱网络以一组来自相关联的肽段的质谱为单位,鉴定出一致性序列,而不是独立地鉴定单个质谱。尽管质谱网络的算法还在发展初期,但它们已经能给出目前最长和最精确的De-novo序列,揭示了一个新的、能发现预计之外的翻译后修饰和高度修饰肽的路径,也使含有未知氨基酸残基的“环状非核糖体肽”的自动测序成为可能,同时定义了一个新的适用于串联质谱分析的、能将生物系统产出的所有分子结果进行映射的方法。
为满足于多个实验数据集的应用,质谱网络库方法所面临的挑战主要来源于数据库的完整性和谱图匹配算法的优化两个方面。
3 质谱网络方法的应用前景
蛋白质的修饰信息是基因组和转录组都无法获得的新的数据,在生物体功能调控等方面具有极其重要的作用,它在质谱数据中体现为某个氨基酸质量的改变[51]。在首次分析可能含有修饰肽的样品时,往往不知道哪些残基或肽被修饰,而质谱图比对方法考虑了每个可能的质谱图对以及匹配质谱之间的每个可能位置的质量差,同时要求匹配的谱峰之间具有显著条件,而不考虑修饰的限制。这种方法可以用于发现新的或非预设的修饰。在实际应用中,Bandeira等[40]使用该方法研究一个 93岁患者的白内障晶状体蛋白质的一组质谱图时,质谱网络不仅能够发现数据库搜索方法鉴定出的修饰,还另外发现了几个新的修饰。
内源肽是机体内存在的天然的生物活性肽,主要包括体内一些重要内分泌腺分泌的肽类激素(如促生长激素释放激素、脾脏中的脾脏活性肽、胰腺分泌的胰岛素等)、由血液或组织中的蛋白质经专一的蛋白水解酶作用而产生的组织激肽(如缓激肽、胰激肽)、作为神经递质或神经活动调节因子的神经多肽以及由昆虫、微生物、植物等生物体产生的抗菌肽。某些神经肽是有价值的治疗靶标[52],某些内源性抗原肽还是免疫治疗策略的关键。深入研究内源肽的 PTMs以及与其相互作用的蛋白质将有助于探索疾病的发生、发展、转移机制等[53]。
目前主要是基于质谱分析方法 內鉴定 源肽[54-55],这种基于仪器识别质谱的方法长期以来试图通过改进仪器和实验方法来降低实验噪声,但由于内源肽的特殊性质,实验提取时不适合消化酶切,尚有大量长肽或短肽以及 PTMs未被鉴定出来。质谱网络方法可以在内源肽的鉴定中发挥重要作用。
同时,蛋白质组学数据的累积为质谱网络方法在大数据分析中提供了广阔的舞台。PRIDE Cluster采用聚类分析了 PRIDE数据库中的全部公开数据,给出了大量含有丰富信息的未鉴定质谱图数据[56],这里面可能包含有大量的PTMs信息,有待我们用类似于质谱网络这样的新方法来进行大数据挖掘。
在质谱网络方法实验过程中,我们利用PRIDE Cluster中未鉴定的高质量质谱数据集,通过谱图库搜索,以及使用PeptideProphet[57]进行质量控制,最终从PRIDE Cluster中获得了很多新的之前未被鉴定的神经肽,该研究成果已经被录用。同时,针对这些未鉴定的肽段,我们使用Spectral Networks工具对该数据集进行分析,找到了38条PTMs的信息。实验表明,基于质谱网络的方法进行谱图库搜索,有利于鉴定未知多肽和翻译后修饰信息。已经报道过的方法中未从谱图层面来实施整个鉴定流程,而是利用数据库搜索方法来鉴定未知肽段,所以使得类似于 PRIDE Cluster这样的数据中存在大量的高质量未鉴定质谱有待于我们研究挖掘。
4 总结
本文主要讨论在蛋白质组学中,基于质谱图比对,建立质谱网络在鉴定多肽翻译后修饰中的发展。来自重叠肽或同一肽的修饰变体的谱图提供了大量相关的序列信息,可以使用基于质谱网络的新一代算法来鉴定。与标准修饰鉴定方法不同,具有来自相同肽的修饰或未修饰变体的谱图允许直接发现样品中的修饰,而不必事先猜测要搜索的修饰列表。来自多种修饰变体的谱图可以组合成质谱网络,并且相关的离子质量和强度可被用于增加鉴定高度修饰的肽的可信度。从蛋白质测序的角度来看,通过非特异性蛋白酶切实现的广泛的序列覆盖可能将来自重叠肽的谱图组装成长的蛋白质重叠群。此外,通过利用组合谱中的相关序列信息,鸟枪蛋白测序方法能够提供有史以来报道过的关于离子阱串联质谱的最高测序准确度。
在复杂生物系统中,蛋白质的精确定量对生物学的许多研究非常重要[58]。而质谱库已被广泛应用于准确研究各种蛋白质定量分析,在定量已知蛋白质的应用研究中尤为突出。这种“定向”方法在研究定向蛋白质组学的过程中允许研究者以多重方式测定数百种蛋白质,该方法在临床和生命科学研究中已被越来越多地使用[59]。
未来我们可以使用未鉴定数据和已鉴定数据来建立质谱网络库,再通过搜库的方法进行质谱的鉴定。这种基于质谱网络库的方法通过建立质谱图对,创建一致性谱等方式可用于鉴定未知质谱的翻译后修饰信息,为质谱图的解析提供了一种新的思路和发展方向。
针对其他内源性多肽修饰(乙酰化、甲基化等)的分析,目前仍需要发展新的方法和技术,面对质谱网络新方法和机器学习、人工智能等技术大量涌现的局面,如数据库搜索方法的改进如何将新方法和新技术标准化并应用也是蛋白质组学鉴定发展面临的问题。
猜你喜欢
美与时代·美术学刊(2022年3期)2022-04-27
食品安全导刊(2021年20期)2021-08-30
现代园艺(2017年13期)2018-01-19
中国公共安全(2017年7期)2017-10-13
摄影之友(影像视觉)(2017年1期)2017-07-18
现代检验医学杂志(2016年3期)2016-11-15
药学与临床研究(2015年4期)2015-06-05
实践·思想理论版(2015年4期)2015-04-16
科学中国人(2015年16期)2015-02-28
特产研究(2014年4期)2014-04-10
