
NiH/Hβ催化剂的制备及催化正己烷异构化性能
2019-02-22 02:41夏道宏陈金射蔡婷婷朱丽君江胜娟段尊斌杨令彬
石油学报(石油加工) 2019年1期
夏道宏, 陈金射, 蔡婷婷, 朱丽君, 江胜娟, 段尊斌, 杨令彬
(中国石油大学(华东) 重质油国家重点实验室, 山东 青岛 266580)
汽油是目前应用最为广泛的发动机燃料之一,是炼油工业的重要产品。辛烷值是汽油质量的重要指标之一。目前,工业上主要通过增加烷基化或重整汽油组分的比例或者添加高辛烷值组分等手段提高汽油的辛烷值[1]。随着炼油工业的发展,轻质正构烷烃通过异构化工艺生产高辛烷值异构烷烃在国内日益受到重视[2]。
目前工业上C5/C6正构烷烃异构化使用的催化剂主要是中温型和低温型贵金属催化剂[3-4],主要缺点是采用价格昂贵的Pt、Pd等贵金属,同时这些催化剂对含硫化合物十分敏感[5],而且低温型异构化催化剂在应用过程中为维持其催化活性需要不断添加卤素,从而会造成设备腐蚀[6-8]。为改进这些缺点,对非贵金属轻质烷烃异构化催化剂的研究受到重视[9]。目前对这类催化剂的研究主要集中在Ni、Mo、Cu等金属组分,关键是如何提高它们在催化剂载体上的分散度,同时提高其催化脱氢/加氢活性,从而提高其催化异构化选择性[9-10]。文献报道,金属氢化物结构独特,其中金属/氢键具有很强的活性,可以进行氧化加成、插入等反应,在多种催化反应中起着关键作用[11-14]。氢化金属具有更高的催化加氢、脱氢功能[11-12,15],若将其应用于催化异构化反应中,可望能够进一步提高催化异构化活性。
笔者主要研究一种具有较好催化异构化性能的金属氢化物催化剂,并探讨氢化金属在催化异构化反应中的作用。以四(三苯氧基磷)氢化镍〔NiH(P(OPh)3)4〕为活性组分前驱体,经负载、还原制备得到NiH/Hβ催化剂,研究其催化正己烷异构化的反应条件,并探讨NiH作为金属活性中心的催化机制。
1 实验部分
1.1 试剂
六水合硝酸镍(AR)、亚磷酸三苯酯(CP)、硼氢化钠(96%)、无水乙醇(AR)以及甲醇(AR)均购自国药集团化学试剂有限公司。Hβ分子筛购自天津南化催化剂有限公司,比表面积690 m2/g,硅/铝原子比25;结晶度大于96.3%,粒径1.5 μm。γ-氧化铝购自临朐县汇泉钼业有限公司,比表面积265 m2/g;粒径45 nm;结晶度大于90%。
1.2 NiH活性组分前驱体NiH(P(OPh)3)4及催化剂的制备
NiH活性组分前驱体NiH(P(OPh)3)4的制备原理见反应式(1)。

(1)
NiH(P(OPh)3)4的制备步骤[16]:首先将亚磷酸三苯酯(0.01 mol)加入到溶有六水合硝酸镍(0.002 mol)的乙醇(20 mL)溶液中;其次将硼氢化钠(0.2 g)溶解在热的乙醇(10 mL)中,并迅速冷却至室温;将硼氢化钠溶液滴入搅拌的上述硝酸镍溶液中,观察到绿色消失,继而产生白色沉淀,继续搅拌10 min,过滤产物并用乙醇洗涤,真空干燥,得到白色粗产物。将粗产物用30 mL加热的苯溶解,趁热过滤,向得到的滤液中加入30 mL甲醇,静置24 h后析出固体,分离得到NiH(P(OPh)3)4。
NiH/Hβ催化剂的制备:将Hβ分子筛与Al2O3粉末按照一定的质量比混合均匀,加入适量的蒸馏水和黏结剂,搅拌均匀,置于TBL-2型催化剂成型机中挤条成型。将成型后的催化剂载体先在红外灯下烤干,研磨成一定粒度的颗粒,再在马福炉中活化。采用等体积浸渍法负载NiH(P(OPh)3)4前驱体,将成形后的分子筛载体浸入NiH(P(OPh)3)4的二氯甲烷溶液中,浸渍时间为24 h,空气中静置直至溶液挥发完全,进一步在N2氛围下80 ℃干燥2 h,制备出NiH(P(OPh)3)4/Hβ。然后采用固定床反应器,将NiH(P(OPh)3)4/Hβ置于反应管中,在H2氛围下反应温度由室温升至300 ℃,原位还原一定时间后,即可将NiH(P(OPh)3)4还原为NiH,制备出NiH/Hβ催化剂。
Ni/Hβ催化剂的制备:为了进行催化异构化性能对比,制备了Ni/Hβ催化剂,其制备方法与NiH/Hβ催化剂类似。以Ni(NO3)2水溶液等体积浸渍上述催化剂载体24 h后,先在空气氛围下120 ℃ 干燥4 h,然后在马福炉中500 ℃焙烧4 h,得到催化剂前驱体NiO/Hβ。随后将其置于固定床反应器中氢气氛围下,温度由室温升至400 ℃还原4 h,得到Ni/Hβ催化剂。
1.3 催化剂的表征方法
样品的物相分析测定在日本理学D/max-ⅢA型X射线衍射仪上进行,光源采用Cu靶Kα辐射,管电压为35 kV,管电流为40 mA,在2θ值为5°~75°的范围内记录谱图,扫描速率为8°/min。样品表面形貌在日本日立公司的X650扫描电镜仪上分析,加速电压为5 kV。红外光谱由Nicolet 6700傅里叶变换红外光谱仪分析,检测器为DTGS检测器。
1.4 催化剂的性能评价
催化剂的活性在高压微反上进行评价。装置所用反应管为550 mm×8 mm的不锈钢管,管外用加热炉加热,管内置热电偶控温。原料正己烷采用无脉冲泵泵入,进料量采用质量流量计计量。反应产物经过分离器分离出液体,进行气相色谱分析。具体操作步骤为:用电子天平称取一定量催化剂置于反应管中间部分,两端用石英砂填满,填装过程中轻轻敲打管外使其在管内紧密。安装好反应管,插上热电偶,通入氢气加压进行气密性检验。待温度稳定后开始进料,反应稳定后开始取样。反应结束后降温,将气体放空,清洗装置备用。
在正己烷的催化异构化反应中,正己烷转化率x、异构烷烃选择性s、异构烷烃质量收率y分别按式(2)、(3)、(4)计算。
x=(∑A-An-C6)/∑A×100%
(2)
s=∑Ai/(∑A-An-C6)×100%
(3)
y=x×s
(4)
其中, ∑A代表色谱图中所有峰的总面积,An-C6代表原料正己烷的峰面积,Ai代表异构化产物中某种异构烷烃的峰面积。
2 结果与讨论
2.1 NiH/Hβ催化剂结构表征
2.1.1 前驱体NiH(P(OPh)3)4的红外光谱分析

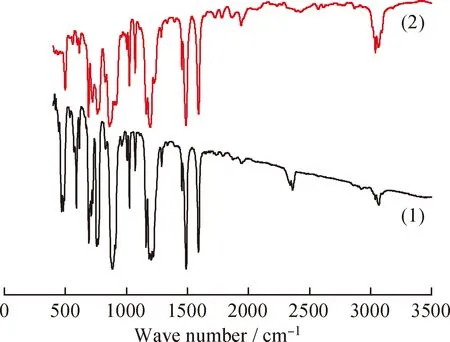
图1 前驱体NiH(P(OPh)3)4和P(OPh)3的红外吸收光谱Fig.1 FT-IR spectra of NiH(P(OPh)3)4precursor and P(OPh)3(1) NiH(P(OPh)3)4; (2) P(OPh)3
2.1.2 前驱体NiH(P(OPh)3)4的XRD分析
催化剂活性组分前驱体NiH(P(OPh)3)4的XRD分析如图2所示。由图2可知,在2θ为8°和18°处有2个明显的衍射峰,表明所合成的NiH(P(OPh)3)4是一种结晶性较强的物质。

图2 前驱体NiH(P(OPh)3)4的XRD谱Fig.2 XRD pattern of NiH(P(OPh)3)4 precursor
2.1.3 载体及NiH(P(OPh)3)4/Hβ的XRD分析
载体和NiH(P(OPh)3)4/Hβ的XRD谱图见图3。由图3可以看出,载体和NiH(P(OPh)3)4/Hβ在2θ为7.94°、21.49°、22.57°、27.17°和29.7°处分别出现归属于Hβ的衍射峰[18],同时在2θ为25.38°和44.02°处出现归属于Al2O3的衍射峰。通过对比发现,NiH(P(OPh)3)4/Hβ的XRD谱图中只出现了载体的衍射峰,而未出现活性组分前驱体NiH(P(OPh)3)4的衍射峰,这说明NiH(P(OPh)3)4在Hβ分子筛上呈现出良好的分散性。

图3 载体和NiH(P(OPh)3)4/Hβ的XRD谱Fig.3 XRD patterns of carrier and NiH(P(OPh)3)4/Hβ(1) Carrier; (2) NiH(P(OPh)3)4/Hβ
2.1.4 载体及NiH(P(OPh)3)4/Hβ的SEM分析
为进一步确定在Hβ分子筛载体上活性组分前驱体NiH(P(OPh)3)4的分散情况,对负载前Hβ载体及负载后NiH(P(OPh)3)4/Hβ的表面形貌进行SEM表征,结果如图4所示。由图4可以看出,Hβ载体与负载后NiH(P(OPh)3)4/Hβ均呈现不规则球形,并且颗粒直径分布不同。同时,在载体表面没有明显观察到前驱体NiH(P(OPh)3)4形成的聚集体,这说明活性组分前驱体NiH(P(OPh)3)4在分子筛载体上负载的较为均匀,与XRD物相分析结果一致。

图4 Hβ载体和NiH(P(OPh)3)4/Hβ的SEM照片Fig.4 SEM images of Hβ carrier and NiH(P(OPh)3)4/Hβ(a) Hβ carrier; (b) NiH(P(OPh)3)4/Hβ
2.2 NiH/Hβ制备条件对催化正己烷异构化性能的影响
本研究主要考察了催化剂的粒度、活性组分含量以及反应条件对其催化异构化活性的影响规律。经证明在本研究的实验条件下,催化异构化反应平衡时间在80~100 min,因此不同因素对催化剂活性影响的数据均为催化反应2 h时(反应平衡后)取样分析的数据。
2.2.1 活性组分负载质量分数对催化剂性能的影响
按照1.2节制备方法,得到活性组分质量分数分别为0.1%、0.15%、0.3%、0.5%、0.7%和0.9%的NiH/Hβ催化剂。
在反应温度300 ℃、压力2.0 MPa、氢/油摩尔比4.0、质量空速1.0 h-1的条件下考察活性组分负载质量分数对催化正己烷异构化反应的影响,结果见图5。从图5可以看出,随着活性组分负载量的增加,正己烷的转化率呈现先上升后下降的趋势。负载质量分数在0.1%~0.5%范围内时,随着其负载质量分数的升高,所制备的催化剂上金属中心数
目增加,有利于正己烷在金属中心上脱氢,正己烷的转化率随之增大。负载质量分数在0.5%~0.9%范围内时,随着活性组分负载质量分数的进一步增加,正己烷的转化率呈下降趋势。这是由于金属中心数目达到一定程度后,正己烷的脱氢反应不再是整个反应的控速步,而此时,活性烯烃生成碳正离子的反应成为控速步,所以,持续增加活性组分的含量反而会堵塞载体的孔道,降低催化剂的活性,导致正己烷的转化率降低。另外,随着NiH负载质量分数的增加,异构烷烃的选择性呈现先下降后升高的趋势。

图5 活性组分负载质量分数(w)对催化正己烷异构化反应性能的影响Fig.5 Effects of NiH mass fraction onn-hexane isomerizationT=300 ℃; p=2.0 MPa; n(H2)/n(n-Hexane)=4.0;MHSV=1.0 h-1
2.2.2 催化剂粒度对催化剂性能的影响
在活性组分NiH负载质量分数为0.2%时,制备出不同粒径大小的NiH/Hβ催化剂,采用与2.2.1节相同反应条件,研究了催化剂粒度对催化正己烷异构化反应性能的影响,结果见图6。由图6可以看出,随着催化剂粒径变大,异构烷烃的收率呈现先升高后降低的趋势。催化剂粒径为0.3 mm时,催化剂的性能相对较好,此时正己烷的转化率为73.8%,异构烷烃选择性及收率分别为82.2%、60.7%。随着催化剂粒径的减小,催化剂的比表面积增大,与正己烷的接触会更加充分,有利于催化剂活性的提高;但当催化剂粒径过小时,正己烷通过催化剂床层时的扩散阻力较大,导致正己烷在催化剂上反应时间过长,加剧了正己烷的裂解程度,最终降低了异构烷烃的收率。
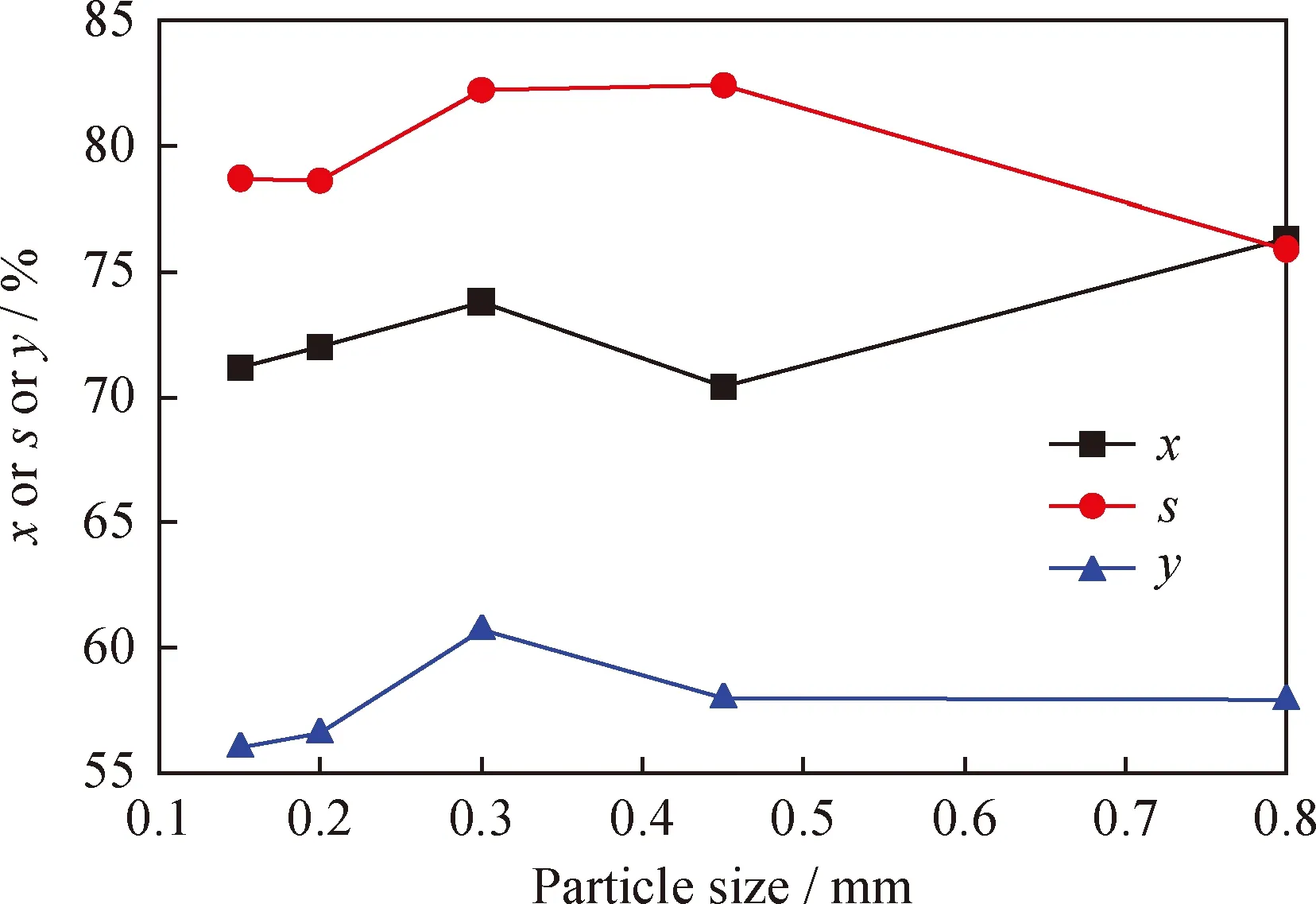
图6 催化剂粒度大小对催化正己烷异构化反应的影响Fig.6 Effects of particle sizes on n-hexane isomerizationT=300 ℃; p=2.0 MPa; n(H2)/n(n-Hexane)=4.0; MHSV=1.0 h-1
2.3 反应条件对催化异构化反应性能的影响
在NiH负载质量分数为0.5%的条件下,原位制备出NiH/Hβ催化剂。考察不同的反应温度、压力、氢/油摩尔比和质量空速对催化剂催化正己烷异构化反应性能的影响,结果见图7。
由图7(a)可知,控制反应压力为2.0 MPa,氢/油摩尔比为4.0,质量空速为1.0 h-1时,随着反应温度的升高,正己烷的转化率呈上升的趋势。
这是因为随着反应温度的升高,反应速率增加,相同时间内转化的正己烷的量增加。然而,异构烷烃选择性的变化趋势与正己烷的转化率正好相反,随着反应温度的升高不断降低。这是由于烷烃在发生异构化反应的同时,伴随着热裂解等副反应的发生,温度的升高同时也增加了副反应的反应速率,从而降低了其选择性。综合考虑正己烷转化率及异构烷烃选择性两方面,催化反应温度以300 ℃为宜。
由图7(b)可以看出,当反应温度为300 ℃、氢/油摩尔比为4.0、质量空速为1.0 h-1时,随着反应压力的增加,正己烷的转化率不断下降。这是由于正己烷在金属中心上脱氢生成烯烃的反应是一个分子数增大的反应,体系压力的升高不利于该反应的发生,从而导致催化异构化转化率降低。异构烷烃的选择性与正己烷的转化率随压力变化的趋势正好相反。这是因为在异构化过程中,如果氢分压过低,活性很高的中间体烯烃分子含量过多,可能会与正碳离子聚合成大分子的烃类,从而降低异构化反应的选择性。综合两方面因素考虑,反应压力选择2.0 MPa。
由图7(c)可以看出,当反应温度为300 ℃、反应压力为2.0 MPa、质量空速为1.0 h-1时,随着氢/油摩尔比的增加,正己烷的转化率缓慢下降。在氢/油摩尔比较低时,正己烷在体系气相中的分压较高,异构化反应的速率较快,使得正己烷的转化率较高;而当氢/油摩尔比较高时,氢气在气相中的分压增大,抑制了正己烷脱氢产生活性烯烃的反应,导致正己烷的转化率降低。异构烷烃的选择性随着氢/油摩尔比的增加而上升。这是由于氢/油摩尔比较低时,尽管会促进正己烷的脱氢反应,但同时生成的活性烯烃会与氢质子结合生成碳正离子或者与正碳离子缩合生成大分子烃类,造成异构烷烃的选择性较低;随着氢/油摩尔比的升高,氢气在气相中的分压增大,有利于生成的活性烯烃较快地进行加氢反应,增加异构烷烃的选择性。综合两方面因素考虑,氢/油摩尔比选择为4.0。

图7 反应条件对NiH/Hβ催化剂催化异构化反应性能的影响Fig.7 Effects of reaction parameters on the performance of NiH/Hβ catalyst(a) p=2.0 MPa, n(H2)/n(n-Hexane)=4.0, MHSV=1.0 h-1; (b) T=300 ℃, n(H2)/n(n-Hexane)=4.0, MHSV=1.0 h-1;(c) T=300 ℃, p=2.0 MPa, MHSV=1.0 h-1; (d) T=300 ℃, p=2.0 MPa, n(H2)/n(n-Hexane)=4.0
由图7(d)可以看出,当反应温度为300 ℃、反应压力为2.0 MPa、氢/油摩尔比为4.0时,随着质量空速的升高,正己烷的转化率降低。质量空速较低时,单位质量催化剂在单位时间内处理的正己烷的量较少,因此,低空速对于提高反应的转化率是有利的,所以此时正己烷的转化率较高;随着质量空速的升高,单位时间内通过催化剂床层的正己烷量增加,部分正己烷还未来得及反应就已经被带离床层,从而导致正己烷的转化率降低。另一方面,随着质量空速的上升,原料在催化剂床层上发生裂解、歧化等副反应的程度减弱,所以异构烷烃的选择性会升高。综合两方面因素考虑,1.0 h-1为最佳质量空速。
为进一步确定催化剂催化正己烷异构化反应的性能,对0.5%NiH/Hβ催化剂催化异构化反应不同时间的转化率及产物分布情况进行了分析,结果见表1。

表1 NiH/Hβ催化剂催化正己烷异构化反应转化率及产物分布Table 1 Conversion and product distribution of n-hexane isomerization on NiH/Hβ
Reaction conditions:T=300 ℃;p=2.0 MPa; MHSV=1.0 h-1;n(H2)/n(n-Hexane)=4.0
由表1数据可以看出,随着反应时间的延长,反应产物中高辛烷值组分2,2-二甲基丁烷及2,3-二甲基丁烷、2-甲基戊烷与3-甲基戊烷的含量逐渐升高,在反应2 h催化异构化反应达平衡后,异构己烷在所有产物中的总质量分数为60.4%,正己烷的转化率达到82.7%,表明该催化剂具有较好的催化异构化性能。
为进行催化异构化性能对比,对只含有金属Ni的0.5%Ni/Hβ催化剂,在同样反应条件下考察其催化正己烷异构化反应的转化率及不同反应时间的产物分布,结果见表2。
对比表1与表2数据可知,不同反应时间Ni/Hβ催化剂催化正己烷转化率均比NiH/Hβ催化剂催化正己烷转化率低;同时,反应2 h时,Ni/Hβ催化正己烷异构化产物中高辛烷值组分2,2-二甲基丁烷及2,3-二甲基丁烷、2-甲基戊烷与3-甲基戊烷的收率均比NiH/Hβ催化正己烷异构化产物低。表明活性组分含有Ni-H的NiH/Hβ催化剂比活性组分不含Ni-H的Ni/Hβ催化剂表现出更高的催化正己烷异构化活性。

表2 Ni/Hβ催化剂催化正己烷异构化反应转化率及产物分布Table 2 Conversion and product distribution of n-hexane isomerization on Ni/Hβ
Reaction conditions:T=300 ℃;p=2.0 MPa; MHSV=1.0 h-1;n(H2)/n(n-Hexane)=4.0
2.4 NiH/Hβ催化轻质烷烃异构化的作用机理
为分析NiH/Hβ催化剂催化轻质烷烃异构化的反应机理,首先对比了H2和N22种气氛下催化剂催化正己烷异构化反应的效果,结果见表3。

表3 N2和H2气氛下NiH/Hβ催化剂催化正己烷异构化反应Table 3 Effects of N2 and H2 on the NiH/Hβcatalytic isomerization of n-hexane
Reaction conditions:T=300 ℃;p=2.0 MPa; MHSV=1.0 h-1;n(H2)/n(n-Hexane)=4.0
由表3可以看出,NiH/Hβ催化剂在N2气氛下催化正己烷异构化反应时,正己烷的转化率和异构烷烃的收率均要比在H2气氛下的低。在烷烃催化异构化反应过程中,烷烃首先在金属中心上发生脱氢反应,经过异构化后生成的异构烯烃再在金属中心上发生加氢反应,最终生成异构烷烃。在N2气氛下,不利于维持金属中心上氢的吸附平衡,导致催化剂的催化加氢活性降低。而在H2气氛下,有利于金属中心上氢的吸附平衡(见图9反应机理),同时H2的存在还会延缓催化剂表面的结焦,维持催化剂的双功能催化活性。
进一步对已制备的NiH/Hβ催化剂进行H2还原处理,处理条件为:H2压力2 MPa,还原温度300 ℃,还原时间2 h。在相同条件下,对进一步还原处理前后的NiH/Hβ催化剂催化正己烷异构化活性进行对比,结果如表4所示。表4结果表明,进一步H2还原处理未能再提高NiH/Hβ催化剂的活性,这说明NiH本身能够有效地催化异构化过程中的加氢/脱氢反应。

表4 进一步氢气还原处理前后NiH/Hβ催化剂的催化活性对比Table 4 Comparison of the catalytic activity ofNiH/Hβ catalysts before and after H2 reduction
Treatment conditions:p=2 MPa; Reduction temperature 300 ℃; Reduction time 2 h
在异构化反应中引发剂所起的作用主要是提供碳正离子,碳正离子的存在可以引发重排反应生成新的碳正离子,使异构化反应容易进行。选取异戊烯、氯代正丁烷、叔丁醇3种较易生成碳正离子的物质做引发剂,考察它们的存在对正己烷异构化反应的影响;氯的存在有可能会增强催化剂的酸性,因此还研究了四氯化碳对该催化剂催化正己烷异构化反应的影响。
在正己烷中分别加入体积分数为0.5%的异戊烯、氯代正丁烷、叔丁醇以及四氯化碳,在反应温度300 ℃、反应压力2.0 MPa、氢/油摩尔比4.0、质量空速1.0 h-1的条件下,分别考察各引发剂对NiH/Hβ催化正己烷异构化反应性能的影响,结果见图8。
由图8可以看出,在异戊烯、氯代正丁烷、叔丁醇做引发剂时,正己烷的转化率和异构烷烃的收率相比于无引发剂时没有太大变化,引发剂所起作用并不明显。这是由于NiH作为催化剂的金属中心,可以迅速地将烷烃脱氢生成活性烯烃,随后烯烃在催化剂的强酸中心上生成碳正离子,所以整个过程无需引发剂生成的碳正离子的引发。由图8还可以看出,原料中加入四氯化碳后,正己烷的转化率和异构烷烃的收率都有大幅度的下降,并且反应完成后催化剂变为绿色,这说明四氯化碳在高温下与NiH作用生成了Ni2+,破坏了催化剂的部分金属活性中心,导致烷烃不能被催化脱氢生成烯烃,所以正己烷的转化率和异构烷烃收率都会下降。
根据实验结果并结合文献报道[19-21],推测NiH/Hβ催化剂催化正己烷异构化的反应机理如图9 所示。反应过程分3步进行:首先2个NiH活性中心共同作用使正己烷脱氢生成烯烃;其次烯烃在催化剂的酸中心上与氢质子结合生成碳正离子,不稳定的仲碳正离子迅速异构化为比较稳定的叔碳正离子,然后脱去氢质子生成带支链的烯烃;第三步是异构化的烯烃在金属中心加氢生成异构烷烃。与传统的双功能异构化催化剂相比,在NiH/Hβ催化剂催化下,正己烷异构化催化脱氢/加氢过程是在金属氢化物活性中心上进行的。
3 结 论
(1)采用浸渍还原法制备了新型的NiH/Hβ异构化催化剂,对催化剂进行表征表明活性组分前驱体NiH(P(OPh)3)4在载体上分布均匀,未发生明显聚集。
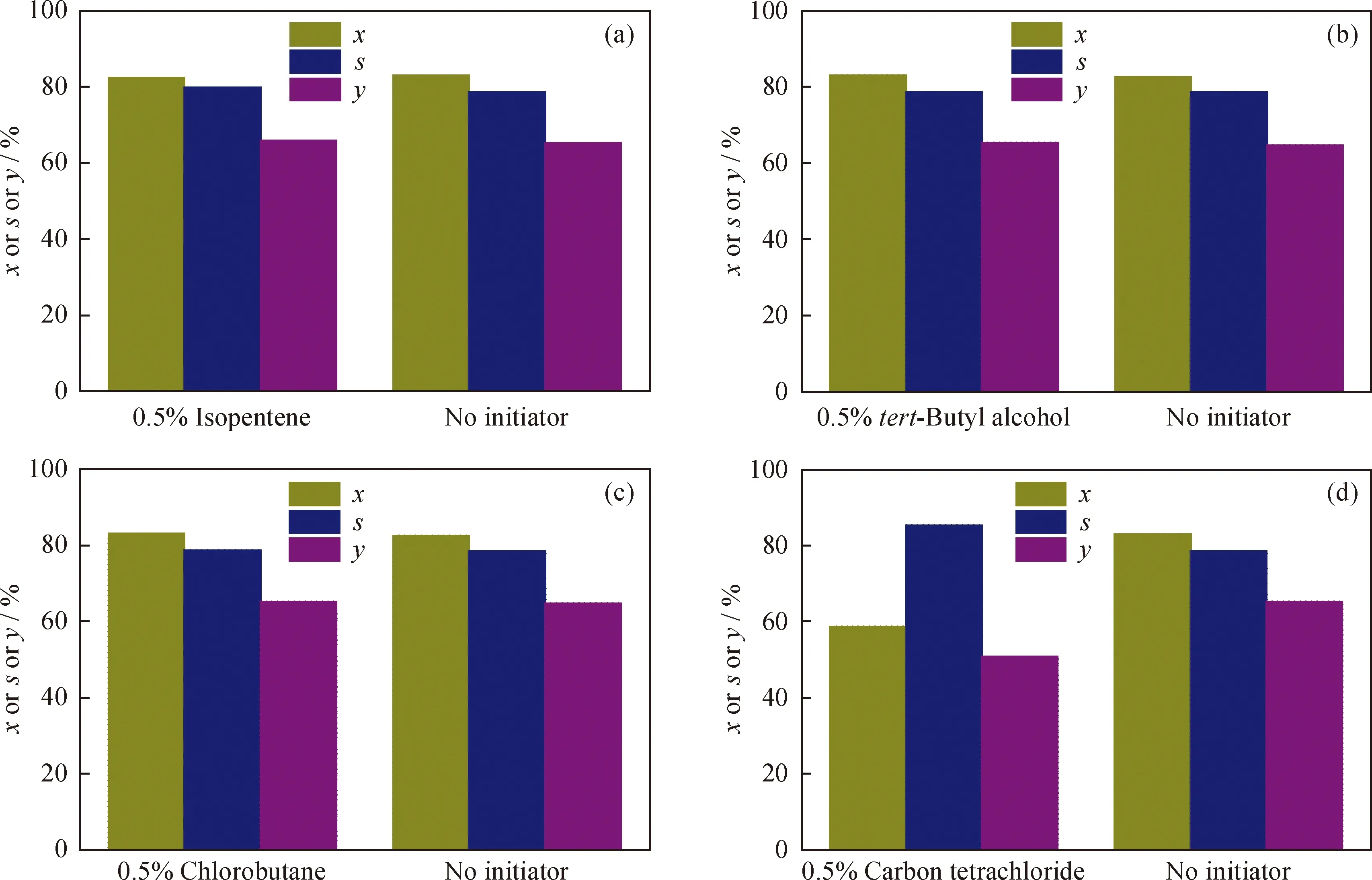
图8 引发剂对NiH/Hβ催化剂催化正己烷异构化反应性能的影响Fig.8 Effect of initiator on NiH/Hβ catalytic n-hexane isomerization(a) Isopentene; (b) tert-Butyl alcohol; (c) Chlorobutane; (d) Carbon tetrachlorideT=300 ℃; p=2.0 MPa; n(H2)/n(n-Hexane)=4.0; MHSV=1.0 h-1

图9 NiH/Hβ催化剂催化正己烷异构化反应机理Fig.9 Mechanism of isomerization of n-hexane catalyzed by NiH/Hβ
(2)考察了NiH/Hβ催化剂制备条件及反应条件对其催化烷烃异构化活性的影响,得到适宜的催化剂活性组分含量以及催化异构化反应条件,即活性组分在载体上的质量分数为0.5%,反应温度为300 ℃,反应压力为2.0 MPa,氢/油摩尔比为4.0,质量空速为1.0 h-1。此时,NiH/Hβ催化剂催化正己烷异构化反应正己烷的转化率为83.0%,异构烷烃的选择性为78.6%,异构烷烃的收率为65.2%。
(3)分析了NiH/Hβ催化剂催化轻质烷烃异构化的反应机理。与传统的双功能异构化催化剂相比,NiH/Hβ催化剂的金属中心为镍氢化物,催化脱氢/加氢过程是在镍氢化物活性中心上进行的,在烷烃异构化反应时具有良好的催化脱氢/加氢功能。
猜你喜欢
化工设计(2022年4期)2023-01-02
纺织标准与质量(2022年3期)2022-08-10
石油炼制与化工(2022年2期)2022-02-15
石油石化绿色低碳(2020年2期)2020-12-31
化工管理(2020年26期)2020-10-09
石油石化绿色低碳(2019年6期)2019-01-14
电子技术与软件工程(2016年24期)2017-02-23
中国石油大学学报(自然科学版)(2015年2期)2015-11-10
应用化工(2015年3期)2015-04-01
中国粮油学报(2014年8期)2014-02-06
