
高能炸药CL-20分子结构的理论模拟方法探究
2019-06-05 02:35魏贤凤
火工品 2019年6期
刘 珉,魏贤凤
高能炸药CL-20分子结构的理论模拟方法探究
刘 珉,魏贤凤
(西南科技大学国防科技学院,四川 绵阳,621010)
采用量子化学方法,在HF/6-31G, DFT-B3LYP/6-31G 和MP2/6-31G 基组水平下全优化ε-CL-20分子的结构,对ε-CL-20的结构(包括键长、键角、二面角)分析得出B3LYP/6-31G水平下的理论值更接近实验值。采用B3LYP 方法,不同基组3-21G,6-31G, 6-31++G, 6-311++G,6-31++G**水平下全优化ε-CL-20分子,基组选取基本不影响计算结果, 表明B3LYP/6-31G 基组水平下的计算结果能够满足ε-CL-20的结构优化要求。采用HF、B3LYP、MP2方法不同基组计算ε-CL-20原子的净电荷分布, 结果表明充分考虑电子相关性的B3LYP 方法计算结果最合理。
炸药;CL-20;量子化学;B3LYP/6-31G;分子结构;HF;MP2
六硝基六氮杂异伍兹烷(HNIW,俗称CL-20)由美国的 Nielsen等[1-2]在 1987年首次合成,是一种最有发展潜力的高能密度化合物之一。CL-20在常温下有4种晶型,即α、β、γ和ε,其中ε-CL-20[3]的分子对称性最高,热力学性能最稳定。量子化学[4]计算方法在预测材料性能方面已得到很大的应用,Hartree-Fock从头计算方法、密度泛函理论(DFT)已被应用于计算晶体的结构和性质[5-7]。肖鹤鸣等人[8]首次运用DFT方法研究CL-20 4种晶型的能带和电子结构,预测感度大小,验证得知ε-CL-20感度最低。
本文运用Hartree-Fock从头计算方法、DFT、MP2方法在6-31G基组水平上对CL-20结构做全优化计算,并在B3LYP方法下采用不同的基组水平对ε-CL-20分子做全优化计算,分析不同计算水平下ε-CL-20的结构和性能,为今后用量子化学方法研究CL-20提供理论支持。
1 计算方法
CL-20常温下有4种晶型,即α、β、γ和ε。主要区别在于它们的硝基在5个氮环平面的取向(即内环和外环之间的差异)上是不同的。其中,ε型热力学性能最稳定,晶型如图1所示。本文采用CL-20最佳几何结构[9],用Gaussian View搭建模型Gaussian09程序全优化计算,采用HF、MP2、B3LYP方法在6-31G水平上全优化分子结构,优化结果收敛正常且无虚频,证明结构是最稳定状态;并采用B3LYP在3-21G、6-31G、6-31++G、6-311++G、6-31++G**水平上全优化计算分子结构,分析优化后的结构和性能。
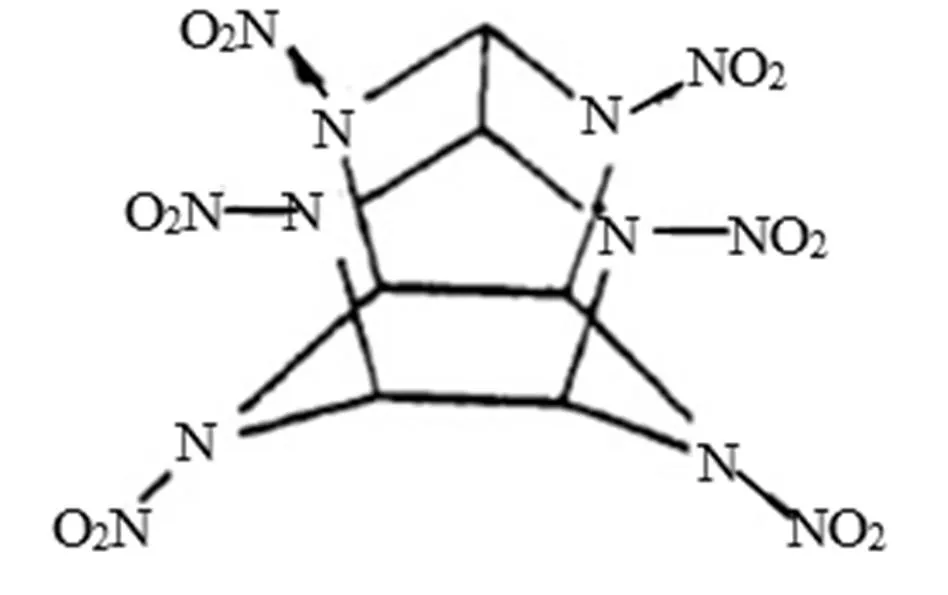
图1 ε-CL-20的晶型
2 结果与讨论
2.1 不同q量化方法优化ε-CL-20结果分析
图2为ε-CL-20分子的原子编号和优化后的几何示意图。由图2可见,CL-20是一多氮多环笼形硝胺分子,其基本结构为一笼形的刚性异伍兹烷,6个硝基分别连接于6个桥氮原子上。
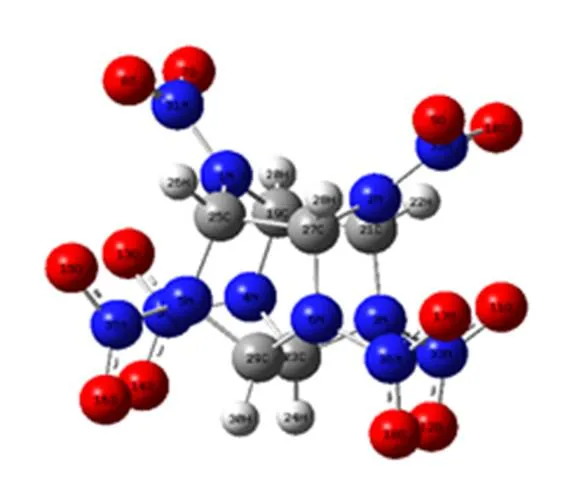
图2 ε-CL-20的结构图
表1~3 分别列出ε-CL-20的全优化几何构型参数键长、键角、二面角,由于此分子结构具有一定的对称性,故表中只列出部分数据。由表1可以看出,HF方法计算的键长比实验值偏短,平均相差0.030nm。MP2方法计算的键长与实验值接近,平均相差0.014nm。B3LYP方法计算的键长也比实验值偏短,平均相差0.001nm。
表1 不同方法在6-31G基组下计算ε-CL-20的键长

Tab.1 The calculated bond lengths of ε-CL-20 at the level of HF/ 6-31G, MP2/ 6-31G and B3LYP/ 6-31G
注:误差值=计算值-参考值
表2 不同方法在6-31G基组下计算ε-CL-20的键角
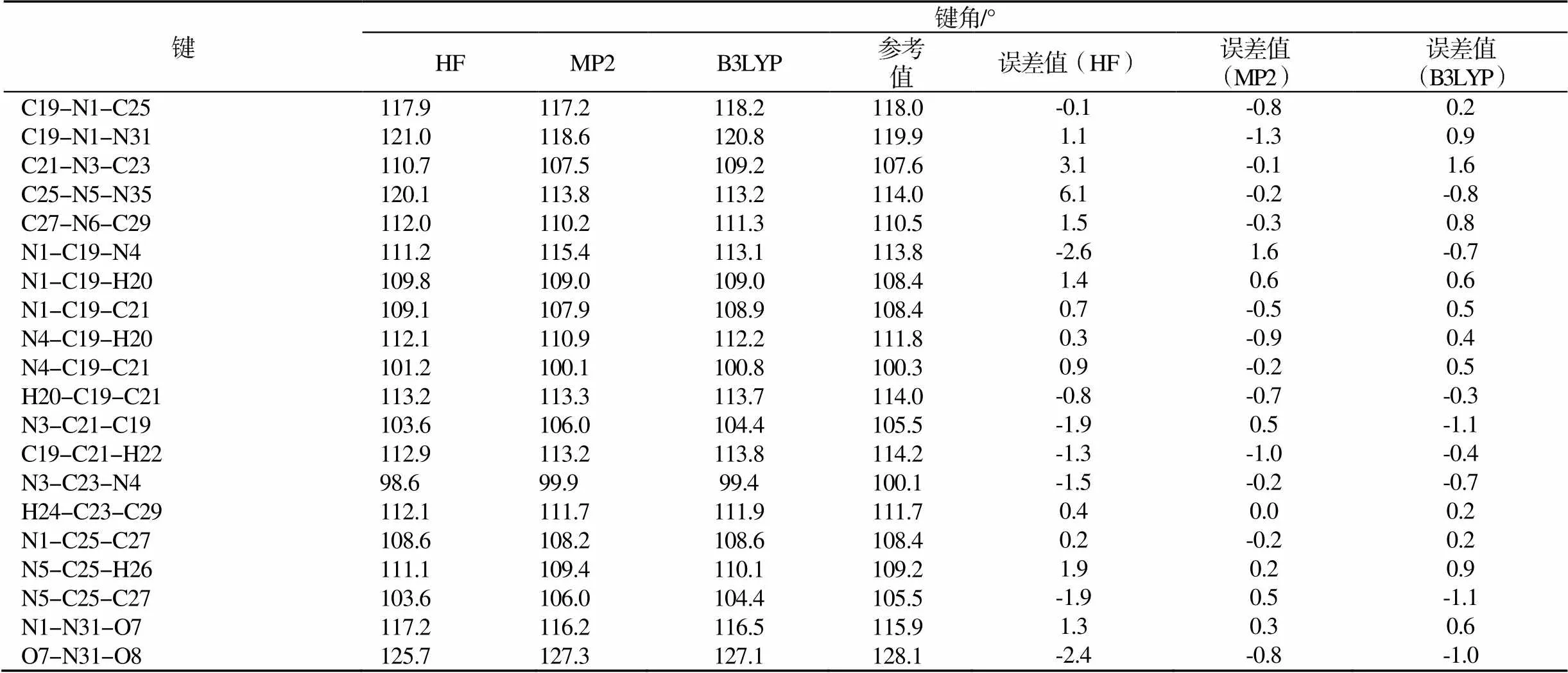
Tab.2 The calculated bond angles of ε-CL-20 at the level of HF/ 6-31G, MP2/ 6-31G and B3LYP/ 6-31G
根据表1中的数据,3种方法计算的键长相差不大,与实验值非常接近。在小基组下,HF只是考虑了电子自旋,而没有考虑电子相关能,所以键长比较短。但是当基组比较小时,HF的键长计算值反而比B3LYP和MP2都精确,这是因为小基组的函数不能满足B3LYP和MP2法的计算要求,所以出现比较大的误差。而且在任何计算方法下,C-H键的键长误差是最小的,而N-N键和N-O键的误差比较大,说明在CL-20分子中,C-H上电子分布比N-N键和N-O键上的电子分布均匀。另外由于C-N单双键长分别是1.471nm和1.273nm,所以C-N键为单键,所有C原子为sp3杂化。而N-N单键长度为1.47nm,N-N双键的长度为1.25nm,所以N-N介于单双键之间。
由表2可以看出,HF方法计算的键角比实验值偏大,平均相差0.659°。MP2法计算的键角与实验值接近,平均相差0.221°。B3LYP方法计算的键角也比实验值偏大,平均相差0.065°。由表2中数据可知,使用3种计算方法对CL-20键角的影响不一样,只有B3LYP方法最接近实验值。其中,以C为中心原子的键角计算值接近于110°,而以N为中心原子的键角计算值接近于119°,由于ε-CL-20分子的中心对称性,可以知道环上的所有C原子采取sp3杂化,环上N原子和硝基N原子均采取sp2杂化。
表3 不同方法在6-31G基组下计算ε-CL-20的二面角

Tab.3 The calculated dihedral angles of ε-CL-20 at the level of HF/ 6-31G, MP2/ 6-31G and B3LYP/ 6-31G
由表3可以看出,HF方法计算的二面角比实验值偏小,平均相差2.4°。MP2法计算的键长与实验值接近,平均相差1.2°。B3LYP方法计算的键长也比实验值偏小,平均相差0.60°。二面角的计算值有一定的偏差,但是MP2方法和B3LYP的计算值更接近,ε-CL-20的分子构型和文献相符。
综上所述,使用MP2,HF,B3LYP这3种方法对ε-CL-20结构进行优化,计算得到的结果都是在实验值误差范围之内。但是,考虑到精度因素,B3LYP方法更接近于实验值,使用B3LYP方法优化ε-CL-20结构更合适。
2.2 不同基组水平下优化ε-CL-20结果分析
基于2.1研究可知采用B3LYP 方法优化出的分子几何构型结果更好。因此,采用B3LYP 方法,取用不同的基组:3-21G,6-31G,6-31++G,6-311++G, 6-31++G**,对结构再次进行优化计算。结果分别见表4~6。
表4 B3LYP方法不同基组下ε-CL-20的计算键长

Tab.4 The calculated bond lengths of ε-CL-20 at different levels with B3LYP method
表5 B3LYP方法不同基组下ε-CL-20的计算键角

Tab.5 The calculated bond angle of ε-CL-20 at different levels with B3LYP method
由表4可以看出,在B3LYP的方法下,除3-21G和6-311++G**基组的计算值以外,其他基组的键长计算值相差都不大。在6-31++G基组下,N-O键的键长与实验值平均相差0.005nm,其他键长基本没有出入。除基组 3-21G和 6-31++G**,其他几个基组的变化不会影响计算的结果,接近于实验值。由表5可以看出,基组的变化对键角的影响不大,以N为中心原子的键角约为120°,以C为中心原子的键角约为110°,都在正常范围内,接近实验值。从表6可以看出,基组的变化对二面角的影响不大,计算出的二面角在正常范围内,因此结构基本不变。
综上所述,相同方法、不同基组计算ε-CL-20分子时结果并没有特别大的差异。但是,与实验值最接近的计算结果是6-311++G基组水平。因为晶体中分子受晶格能束缚和周围分子的影响,而且计算的结果为理想气体状态下的分子结构,所选用的基组也有一定近似性,所以,优化计算所得构型与晶体状态下的分子结构不可能完全一致,计算值与晶体数据之间是存在一定偏差的。因此综合考虑,选取B3LYP/6-31G优化的结构基本可以满足需求。
表6 B3LYP方法不同基组下ε-CL-20的计算二面角

Tab.6 The calculated dihedral angle of ε-CL-20 at different levels with B3LYP method
2.3 CL-20的计算时间和单点能分析
不同方法全优化-CL-20分子的时间图见图3。

图3 不同方法优化ε-CL-20分子的时间柱形图
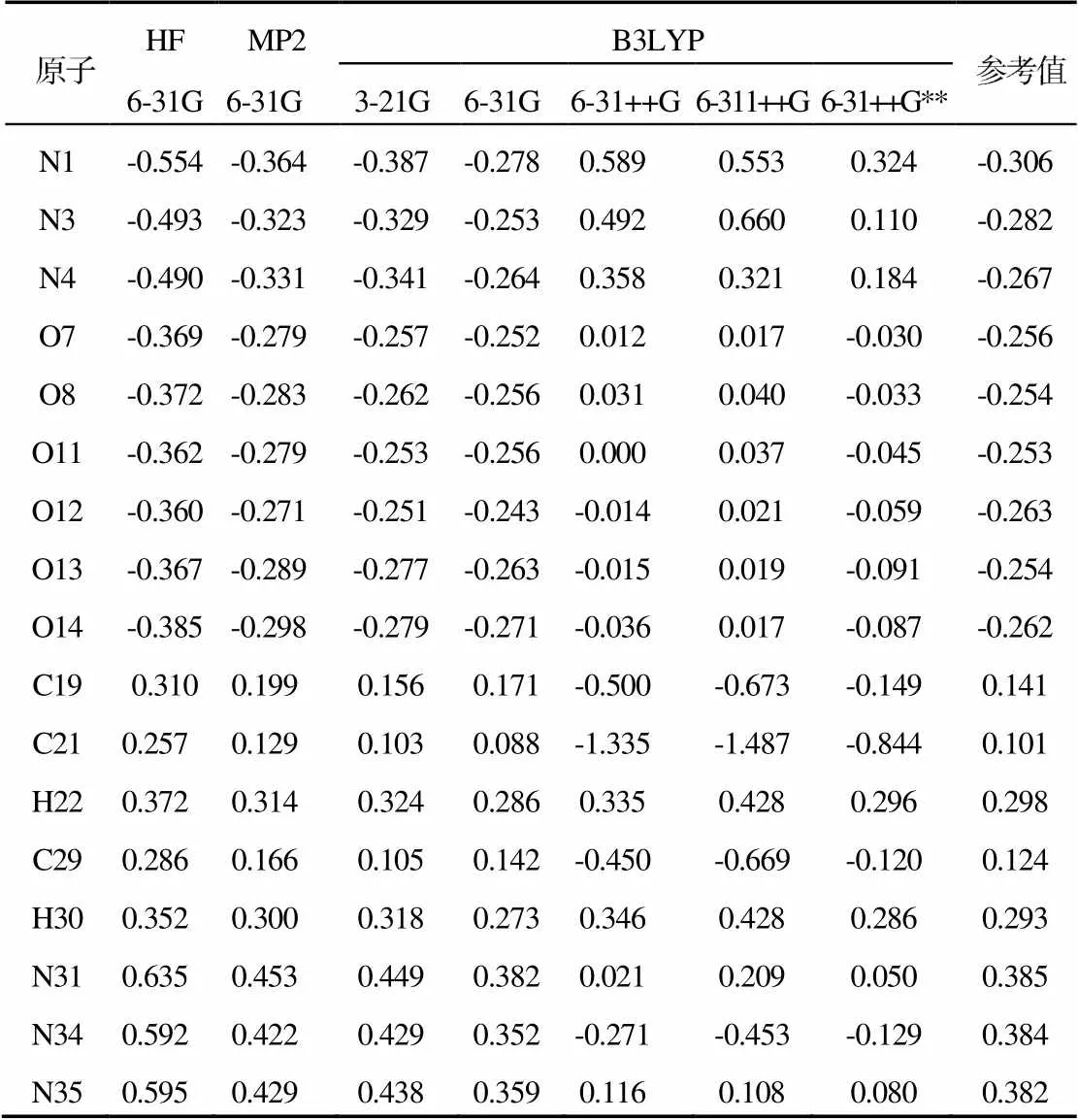
由图3可以看出,在相同基组、不同方法下,计算的时间长短顺序为HF 不同方法不同基组下CL-20的单点能计算如表7所示。 表7 不同方法计算CL-20的单点能(hartree) Tab.7 The calculated single point energy of ε-CL-20 with different methods 由表7可以看出,不同方法不同基组下CL-20的能量相差不大,与实验值一致。HF法因为没有考虑电子相关,MP2法只考虑了60%的相关能,整个计算结果的能量都偏低[10]。在B3LYP方法中3-21G基组与其他基组计算结果相差较大,而其他基组呈现出基组越大,能量越大的趋势。这是因为大基组加入了极化、弥散等条件,分子中更多的能量被考虑到,而且大基组分为多个基函数,使误差降到更低[11]。综上所述,小基组的计算值偏小,大基组考虑了更多的相关能后,能量增加,总体来说CL-20的能量变化不大,都在误差允许范围内。通过比较HF、MP2、B3LYP与实验值,发现B3LYP的计算结果最接近实验值。而采用B3LYP方法不同基组3-21G、6-31G、 6-31++G、6-311++G、6-31++G**水平下,基组越大,计算精度越高,但时间也越长。综合考虑B3LYP/ 6-31G为ε-CL-20结构计算的最优方法。 电荷集居数分布是由电子波函数决定的分子中的电子在各原子上和原子间的分布密度。通过不同的方法、不同的基组计算了ε-CL-20上各原子电荷的分布情况。由于电荷分布具有中心对称性,表8中只列出了部分原子的电荷分布。 由表8可以看出,通过不同的方法、不同基组计算ε-CL-20上各原子电荷的分布情况差别较大。在同一基组(6-31G)、不同方法水平下,亚硝基上的N原子带正电荷,O原子带负电荷且相差不大,H原子带正电荷,环上的C原子带正电荷,环上的N原子带负电荷。 采用B3LYP方法计算不同基组3-21G、 6-31G、 6-31++G、 6-311++G、6-31++G**水平下的原子净电荷。除3-21G和 6-31G基组外,其它基组的计算结果是环上的C原子带负电荷,环上的N原子为正电荷,这与N原子上的分析结果和NO2有很大的不同。电荷不规则,O原子电荷接近0。因此,采用B3LYP方法在 3-21G、 6-31++G、 6-311++G和 6-31++G**基组水平下均不适合计算净电荷的分布。基组3-21G太小,会产生较大的误差;而基组 6-31++G、 6-311++G、6-31++G**可能是因为增加了极化和弥散函数使结果异常;基组6-31G的计算结果最合理。 总之,采用不同方法相同基组与参考值的比较,得出不同方法的精度顺序从低到高依次为HF、MP2、B3LYP;而在B3LYP方法不同基组水平下分析计算结果得出基组6-31G计算准确。 表8 不同水平下ε-CL-20原子电荷的分布 Tab.8 The net charges distribution of ε-CL-20 atoms at different levels (1)对于高能炸药ε-CL-20,充分考虑电子相关性的B3LYP方法计算的结果比HF方法计算的结果要更准确,并且耗费的时间比MP2要少。(2)综合考虑时间和计算精度两方面因素,用B3LYP/6-31G 水平基本上就可以满足对ε-CL-20 结构优化和单点能计算的要求。 [1] Nielsen A T.Cagedpolynitramine compound:US,5693794 [P]. 1997-12-02. [2] 李文祥,赵省向,邢晓玲.CL-20的合成、性能和应用研究进展[J].山西化工, 2015, 35(2):28-30. [3] 文国. CL-20晶型研究及CL-20/TATB共晶的分子动力学模拟[D].太原:中北大学, 2014. [4] 龙威.理论研究中的量子化学计算方法[J].宁夏师范学院学报, 2010,31(3):43-47. [5] Zhu W H,Xiao J J,Xiao H M.Comparative first-principles study of structural and optical properties of alkali metal azides[J]. J Phys Chem B, 2006(110):9 856-9 862. [6] Zhu W H,Xiao J J,Xiao H M.Density functional theory study of the structural and optical propertirs of lithium azide[J].Chem Phys Lett,2006(422):117-121. [7] Zhu W H,Xiao H M.Ab. Initio study of energetic solids:cupric azide,mercuric azides,and lead azide[J].J Phys Chem B,2006(110):18 196-18 203. [8] Xu X J,Zhu W H,Xiao H M.DFT studies on the four polymorphs of crystalline ε-CL-20 and the influences of hydro- static pressure on CL-20 crystal[J].J Phys Chem B,2007(111):2 090-2 097. [9] 赵信歧,施倪承.ε-六硝基六氮杂异伍兹烷的晶体结构[J].科学通报,1995(40):2 158-2 160. [10] Zhu K,Achord P D, Zhang X, et al. Highly effective pincer-ligated iridium catalysts for alkane dehydrogenation DFT calculations of relevant thermodynamic, kinetic, and spectroscopic properties.[J]. Journal of the American Chemical Society, 2004, 126(40):13 044-53. [11] Ciofini I, Daul C A. DFT calculations of molecular magnetic properties of coordination compounds[J].Coordination Chemi- stry Reviews, 2003, 238(02):187-209. Study on Methods for Theoretical Modeling the Structure of High Explosive CL-20 LIU Min,WEI Xian-feng (Southwest University of Science and Technology, Mianyang, 621010) Quantum chemistry methods were used to optimize the structure of ε-CL-20 at the level of HF/6-31G, DFT-B3LYP/6-31G and MP2/6-31G respectively. The structure of ε-CL-20, such as bond length, bond angle, dihedral angle,was analyzed theoretically.The theoretical value at the level of B3LYP/6-31G accords with the experimental value. The effects of B3LYP/3-21G, B3LYP/6-31G, B3LYP/6-31++G, B3LYP/6-311++G and B3LYP/6-31++G** methods on the calculated results of ε-CL-20 are small, which shows that the calculated results at the level of B3LYP/6-31G can meet the optimization of ε-CL-20. The atom charges distribution of ε-CL-20 at different levels were calculated quantitatively. The results show that the B3LYP method in which electron correlation effect has been sufficiently considered is more reasonable. Explosives;CL-20;Quantum chemistry;B3LYP/6-31G;Molecular structure;HF;MP2 TQ564 A 10.3969/j.issn.1003-1480.2019.06.007 1003-1480(2019)06-0025-05 2019-09-17 刘珉(1986 -),女,硕士研究生,主要从事火工品设计、含能材料模拟研究。 中国国家自然科学基金会财政支持(项目批准立项号11602239)。
2.4 CL-20的电荷集居数(Mulliken)的分析
3 结论
猜你喜欢
中学生数理化(高中版.高考数学)(2022年2期)2022-04-26
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
中学生数理化(高中版.高考数学)(2020年3期)2020-05-25
高中生·天天向上(2018年1期)2018-04-14
小天使·三年级语数英综合(2017年6期)2017-06-07
小天使·三年级语数英综合(2017年6期)2017-06-07
中学生数理化·高三版(2017年3期)2017-04-21
数理化学习·高三版(2015年3期)2015-10-21
