
高通量测序解析微生物肥料对红壤有机农田土壤微生物群落的影响
2019-08-13 08:54王超陈刘军田伟
江苏农业科学 2019年2期
王超 陈刘军 田伟
摘要:高通量测序技术可用于精确分析土壤微生物群落,从微生物群落结构和多样性的角度阐释微生物肥料对有机农田根区土壤微生物群落的影响。在红壤有机农田轮作种植条件下,施用微生物肥料后利用Illumina MiSeq高通量测序技术结合相关生物信息学分析土壤细菌和真菌的多样性指数及群落结构。结果表明,从6个有机农田根区土壤样本中获得7 729个细菌分类操作单元(operational taxonomic units,简称OTU)和3 271个真菌OTU,细菌和真菌文库测序覆盖率均在99%以上。微生物肥料会显著降低土壤细菌和真菌种群多样性,且可在一定程度上降低细菌群落丰富度,显著降低真菌群落丰富度;并减少根区土壤特有细菌和真菌物种数量。放线菌门、变形菌门和酸杆菌门是优势细菌,子囊菌门是优势真菌;微生物肥料会提高放线菌门、变形菌门、厚壁菌门和担子菌门的相对丰度,分别比对照组提高29.46%、9.17%、129.33%、165.73%;但会降低酸杆菌门、绿弯菌门、子囊菌门、接合菌门的相对丰度,分别比对照组降低30.14%、33.50%、17.27%、86.33%。因此,施用微生物肥料可改变红壤有机农田根区土壤细菌和真菌的丰富度和多样性,有助于控制作物病害发生。
关键词:微生物肥料;根区土壤;Illumina Miseq高通量测序;微生物群落;种群多样性;相对丰度
中图分类号: S144 文献标志码: A 文章编号:1002-1302(2019)02-0272-05
健康的土壤是农业种植生产的基础,通过施肥培育健康土壤、提升土地质量是当前我国农业种植生产中提高作物产量和品质的关键环节。与常规农业中化学肥料的大量使用相比,施用生物有机肥及微生物肥料是当前有机种植生产中改良土壤、增加土壤肥力的重要手段。微生物肥料中含有特定功能的微生物,可以调控农作物的生长发育,减少或降低作物病虫害的发生,改善农产品品质[1]。近年来,我国微生物肥料产业快速发展,截至2016年7月,农业部登记的微生物肥料产品达到2 780个,年产量已突破1 000万t,应用面积超过1 333万hm2[2]。与之相对应的是我国有机作物种植面积的快速增长,截至2015年年底,有机植物生产面积为222.4万hm2,有机生产面积已达全国农业耕地面积的0.77%[3]。随着我国有机作物种植面积的不断增加,微生物肥料的施用量增加,然而微生物肥料在有机种植耕作层土壤质量提升中的作用尚缺乏明确的科学依据。微生物肥料可分为根瘤菌肥料、固氮菌肥料、硅酸盐细菌肥料、磷细菌肥料和复合微生物肥料,有效活菌数要求≥1亿个/mL[1]。田间施用微生物肥料会将大量微生物引入土壤局部区域,对有机种植土壤中原有的微生物群落造成影响,甚至会破坏土壤微生态平衡。因此,须要科学评测微生物肥料对有机农田土壤微生物群落的影响。
高通量测序技术是目前应用最普遍的新一代测序技术,与传统的琼脂培养基培养、Biolog平板法、磷脂脂肪酸、变性梯度凝胶电泳(polymerase chain reaction-denaturing gradient gel electrophoresis,简称PCR-DGGE)和限制性片段长度多态性(restriction fragment length polymorphism,简称RFLP)等分析方法相比,具有高通量、高灵敏度、高准确性和低运行成本等特点,通过检测土壤微生物细胞内特定遗传物质(原核微生物16S rRNA、真核微生物18S rRNA)的碱基序列,可以更全面、更准确地揭示土壤中微生物群落的复杂性和多样性,已被广泛应用于土壤微生物群落研究中[4-6]。本研究基于Illumina新一代MiSeq平台的高通量测序技术,结合相关生物信息学方法,全面分析土壤细菌16S rRNA基因V3+V4区和真菌ITS1区的多样性指数及群落结构,旨在研究云南地区红壤有机农田在轮作种植条件下,微生物肥料对有机农田根区土壤微生物多样性和群落结构的影响,以期科学地反映施用微生物肥料对有机种植土壤健康的影响。
1 材料与方法
1.1 试验地概况
田间试验选址在云南芸岭鲜生农业发展有限公司庄科基地有机蔬菜田(102°52′33.8″E,25°13′53.7″N),土壤类型为红壤,年平均气温为14.9 ℃,年日照时数为2 205.1 h,无霜期为278 d,年均降水量约为1 000.5 mm。经本研究种植前采样分析测定得出,试验田0~20 cm土壤基本性质:pH值为 6.25,铵态氮含量为27.27 mg/kg,速效磷含量为 37.53 mg/kg,速效钾含量为342.06 mg/kg,有机质含量为33.37 g/kg。
1.2 试验设计
试验于2017年5—10月进行,采用随机区组设计,共设置2个处理,分别为微生物肥料处理(N)和对照(C),每个处理设置3次重复,共6个小区,每个小区面积为3 m×10 m=30 m2,保护行宽为1 m,走道宽及小区间排水沟宽均为 0.5 m。试验作物为辣椒(Capsicum annuum L.),前茬作物为青菜(Brassica chinensis L.)。微生物肥料处理组于辣椒定植后和盛花期分别在根部施用微生物肥料的200倍稀释液,对照组施用等量灌溉水。田间管理及有害生物防治采取有机种植模式。
微生物肥料由南京農大生物源农药创制有限公司提供,已通过南京国环有机产品认证中心有机农业生产资料评估(证明号:IP-0109-932-1696),被广泛应用于江苏等地区的有机种植;登记证号为微生物肥(2013)准字(1096)号,有效菌种为枯草芽孢杆菌(Bacillus subtilis),有效活菌数≥2.0亿个/mL,pH值为3.5~5.0。
1.3 土壤样品采集
土壤样品采集于盛果期进行,用土壤采样器钻取各处理小区0~20 cm根区的土壤,按照“S”形多点取样,每个小区取6钻土壤,去除根系、杂草、土壤动物和石块等杂质后充分混匀作为1个根区土壤样品,采用四分法将其平均分成2份,低温保存带回实验室,置于-80 ℃冰箱保存备用。
1.4 土壤湿化性质测定
土壤样品经自然风干、研磨过筛(孔径为1 mm)后,参照文献[7]中的方法测定土壤理化性质。其中,pH值使用pH计测定(土水体积比为1 ∶ 5);土壤样品经浓度为0.5 mol/L的K2SO4溶液浸提1 h后,用连续流动分析仪测定氨态氮含量;速效磷含量采用0.5 mol/L NaHCO3浸提-分光光度法测定;速效钾含量采用1 mol/L乙酸铵浸提-火焰光度法测定;有机质含量采用油浴外加热-K2Cr2O7容量法测定。
1.5 土壤微生物总DNA提取和高通量测序
采用E.Z.N.A. Soil DNA Kit(OMEGA,美国)试剂盒提取土壤微生物总DNA,采用NanoDrop-ND1000测定提取的DNA浓度,并经2%琼脂糖凝胶电泳对DNA样品进行检测,合格后用于构建文库;以各土壤样品微生物总DNA为模板,以细菌V3+V4区特异性引物(338F 5′-ACTCCTACGGGAG GCAGCAG-3′;806R 5′-GGACTACHVGGGTWTCTAAT-3′)[8]和真菌ITS1区特异性引物(ITS1F 5′-CTTGGTCATTTA GAGGAAGTAA-3′;2043R 5′-GCTGCGTTCTTCATCGATG C-3′)[4]进行PCR扩增;采用Illumina MiSeq测序平台对PCR扩增产物进行双端测序分析,委托上海美吉生物医药科技有限公司完成测序。
1.6 数据处理与分析
测序数据通过FLASH[9]和Trimmomatic[10]软件进行过滤优化和双端序列的连接,优质序列利用Usearch软件基于97%的相似水平进行分类操作单元(operational taxonomic units,简称OTU)聚类,根据Silva细菌数据库[11]和Unite真菌数据库利用RDP Classifier进行物种注释和分类[12];基于OTU丰度信息,利用R语言工具进行主成分分析(principal component analysis,简称PCA),制作稀释曲线(rarefaction curve)、Venn图和群落柱形图;利用Mothur软件计算α-多样性指数(Shannon、Simpson、Chao、ACE等指数)[5,13],并用Excel和DPS软件进行数据分析。
2 结果与分析
2.1 土壤微生物文库测序结果评价
本研究对6个土壤样品进行了Illumina MiSeq高通量测序,数据经过滤优化,细菌文库共得到296 292条有效序列,序列长度分布在256~532 bp之间,在97%相似水平上聚类获得7 729个OTU;真菌文库共得到429 870条有效序列,序列长度分布在202~456 bp之间,在97%相似水平上聚类获得3 271个OTU。细菌V3+V4区、真菌ITS1区的有效序列数量和OTU数量如表1所示,处理组和对照组细菌文库的有效序列数量和OTU数量差异不显著;处理组真菌文库的OTU数量显著低于对照组,两者有效序列数量差异不显著。
稀释曲线反映了样品文库测序数据量的合理性,可用于评价测序数据能否覆盖所有类群。細菌和真菌多样性稀释曲线(图1)显示,随着测序量的不断增大,各样品OTU数目的增加趋势趋于平缓(基本达到饱和),说明测序数据量合理。在97%相似水平上计算各土壤样品测序的覆盖率,由表2可知,细菌、真菌文库测序覆盖率均在99%以上,说明取样合理,处理组间微生物文库测序覆盖率差异不显著。因此,测序数据能够真实地反映土壤样品中的微生物群落,但可能仍有少量微生物种类未被发现。
2.2 微生物肥料对根区土壤微生物群落丰富度和多样性的影响
Shannon指数、Simpson指数用于反映土壤样品中微生物的多样性,前者数值越大,表明群落多样性越高;后者数值越大,表明群落多样性越低[14]。由表2可知,与对照组相比,施用微生物肥料后,有机农田根区土壤细菌Shannon指数显著降低,Simpson指数显著升高,幅度分别为6.84%、170.31%;土壤真菌Shannon指数显著降低,Simpson指数显著升高,幅度分别为30.57%、125.08%。可见,有机农田根区土壤施用微生物肥料会显著降低细菌和真菌的种群多样性。
ACE指数、Chao指数可反映群落物种丰富度。由表2可知,有机农田根区土壤细菌ACE指数、Chao指数在施用微生物肥料后均有所降低,幅度分别为2.76%、2.05%,与对照组差异不显著;与对照组相比,施用微生物肥料可以显著降低真菌的ACE指数、Chao指数,分别降低29.28%、35.57%。结果表明,有机农田根区土壤施用微生物肥料可在一定程度上降低细菌群落丰富度,但显著降低真菌群落丰富度。
2.3 施用微生物肥料后根区土壤微生物类群分析
由表1可知,在97%相似度水平上,对照组、处理组土壤样品分别得到1 309、1 267个细菌OTU,两者差异不显著;分别得到685、405个真菌OTU,对照组显著高于处理组。可见,微生物肥料可在一定程度上降低根区土壤微生物类群的特异性。
Venn图可直观展现并反映组间或样品之间OTU数量组成相似性、重叠情况以及特异性[15]。由图2-A可知,对照组和处理组之间共有的细菌OTU数量为1 300个,代表物种(占比超过1.5%的纲)分别属于放线菌纲(Actinobacteria)和α-变形菌纲(Alphaproteobacteria)。对照组、处理组特有的OTU数量分别为41、26个,说明施用微生物肥料会减少根区土壤特有细菌物种。从图2-B可以看出,对照组和处理组共有的真菌OTU数量为456个,主要共有物种(占比超过5%的纲)分别属于银耳纲(Tremellomycetes)和粪壳菌纲(Sordariomycetes)等。对照组、处理组特有的OTU数量分别为334、21个,说明施用微生物肥料会减少根区土壤特有真菌物种。
PCA是一种对数据进行简化分析的技术,通过分析不同样本群落组成,反映样本间的差异和距离,如样本物种组成越相似,反映在PCA图中的距离越近[16]。在97%相似度水平上,对不同处理组土壤样品细菌OTU组成进行主成分提取,方差最大化正交旋转后(P=0.05)提取了2个主成分,贡献率分别为75.51%、10.57%(图3-A),处理组和对照组的样本围绕主成分1轴完全分开,分别分布在主成分1轴的正轴、负轴,说明处理组和对照组根区土壤细菌群落结构差异明显。真菌OTU PCA获得了贡献率分别为81.59%、8.30%的2个主成分(图3-B),处理组的3个样本分布在主成分1轴的正轴,对照组分布在主成分1轴的负轴,表明处理组和对照组根区土壤真菌群落结构差异明显。
2.4 微生物肥料对根区土壤微生物群落分布特征的影响
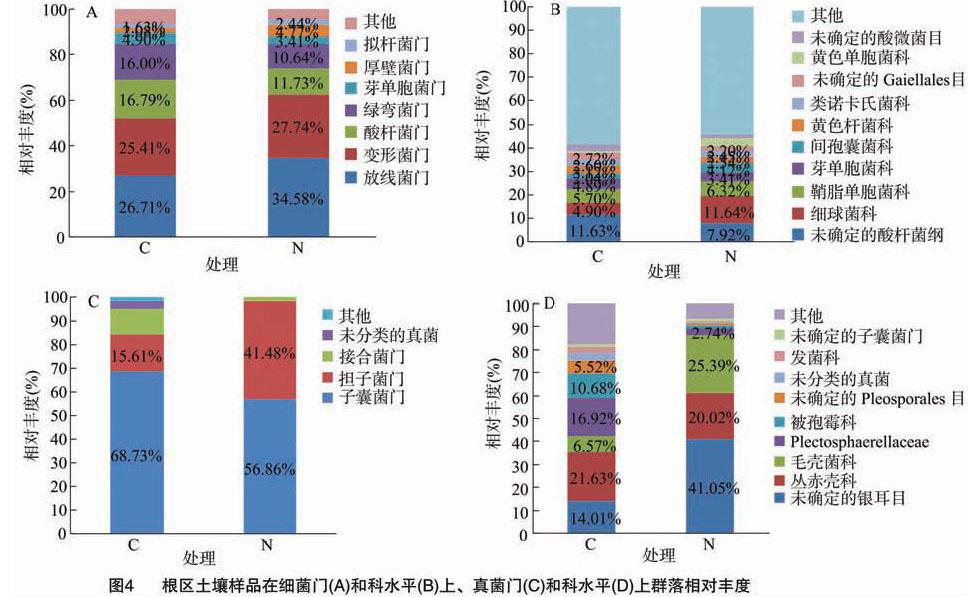
在门分类水平上的细菌类群分布及相对丰度如图4-A所示,有机农田根区土壤样品中的细菌包含放线菌门(Actinobacteria)、变形菌门(Proteobacteria)、酸杆菌门(Acidobacteria)、绿弯菌门(Chloroflexi)、芽单胞菌门(Gemmatimonadetes)、厚壁菌门(Firmicutes)、拟杆菌门(Bacteroidetes)等菌群,处理组和对照组细菌群落组成相似;除未分类的其他菌群外,放线菌门、变形菌门和酸杆菌门的相对丰度较高,属于优势细菌;施用微生物肥料可不同程度地提高放线菌门、变形菌门和厚壁菌门的相对丰度,分别比对照组提高29.46%、9.17%、129.33%;降低根区土壤酸杆菌门、绿弯菌门和芽单胞菌门的相对丰度,分别比对照组降低30.14%、
33.50%、30.41%。从细菌科的分类水平上(图 4-B)看,除未确定科类外,有机农田根区土壤中细球菌科(Micrococcaceae)、鞘脂单胞菌科(Sphingomonadaceae)和芽单胞菌科(Gemmatimonadaceae)的相对丰度均较高(相对丰度>3%),属于优势细菌;施用微生物肥料可使芽单胞杆菌科的相对丰度降低30.27%,细球菌科、鞘脂单胞菌科的相对丰度分别升高137.55%、10.88%。
图4-C为根区土壤真菌在门分类水平上的类群分布及相对丰度,子囊菌门(Ascomycota)、担子菌门(Basidiomycota)、接合菌门(Zygomycota)等在处理组和对照组土壤样品中均有分布,其中子囊菌门是优势菌群,相对丰度占比超过56%;受限于数据库的原因,相对丰度排名第4的菌群未能分类;施用微生物肥料使子囊菌门、接合菌门的相对丰度分别降低 17.27%、86.33%,担子菌门的相对丰度提高165.73%。由图4-D可知,有机农田根区土壤真菌在科水平上主要包括丛赤壳科(Nectriaceae)、毛壳菌科(Chaetomiaceae)、被孢霉科(Mortierellaceae)等;施用微生物肥料可提高毛壳菌科的相对丰度,比对照组提高286.45%,降低丛赤壳科和被孢霉科的相对丰度,分别比对照组降低 7.44%、86.61%。
3 讨论与结论
当前,高通量测序技术已被广泛用于土壤微生物的群落结构及多样性研究中[6]。针对外源有益微生物对有机农田土壤微生态影響的研究鲜有报道,本研究基于Illumina MiSeq平台,对云南地区红壤有机农田根区土壤细菌16S rRNA基因V3+V4区域和真菌ITS1区域进行高通量测序,结合相关生物信息学方法分析发现,处理组和对照组根区土壤微生物群落结构差异明显,微生物肥料会显著降低土壤真菌群落多样性和丰富度,与Zhao等在西瓜种植中应用菌肥的研究结果[17]一致。基于Silva、Unite数据库分析显示,在有机农田根区土壤中,放线菌门、变形菌门、酸杆菌门等是主要细菌类群,与杨亚东等针对不同类型农田土壤分析得到的细菌优势类群相似[18-19],表明农田土壤中细菌的优势类群相似;子囊菌门、担子菌门、接合菌门等是主要真菌类群,与陈丹梅等对云南植烟红壤土壤微生物群落的研究结果[20]一致。
α-多样性指数是有效评价土壤微生物群落多样性和物种丰富度的指标[21]。本研究发现,施用微生物肥料显著降低了有机农田根区土壤细菌多样性Shannon指数,丰富度ACE指数、Chao指数分别比对照组降低2.76%、2.05%,与以往研究得出的植物根际促生细菌(plant growth-promoting rhizobacterium,简称PGPR)能提高土壤细菌种群多样性的结论[22-23]不一致,可能因为微生物肥料中枯草芽孢杆菌的拮抗作用影响了其他土壤细菌的生存。大多植物病原菌为真菌,康捷等发现,麻山药糊头病和根茎腐病发病植株土壤真菌多样性和丰富度均高于健康植株土壤[24];本研究结果显示,微生物肥料能显著降低有机农田根区土壤真菌多样性和丰富度,与Luo等的研究结果[25]一致,表明微生物肥料会在一定程度上减少有机农田作物受病原真菌侵害的几率。枯草芽孢杆菌是目前研究较多的生防细菌之一,对多种植物病原菌具有抑制作用[26],本研究选用的微生物肥料也已被证实对作物真菌病害具有防治效果[27-28]。
植物对土传病害的抗性与根际土壤微生物群落结构密切相关,抗病品种根际土壤放线菌数量和比例明显高于感病品种[29]。姜飞等发现,嫁接辣椒植株根际土壤放线菌比例增加是其根腐病抗性增强的重要原因[30]。本研究同样发现,施用微生物肥料后,处理组根区土壤放线菌门的相对丰度比对照组提高29.46%;同时发现,施用微生物肥料会提高根区土壤真菌毛壳菌科的相对丰度。施用玉米秸秆生物炭后,黄瓜结果期土壤子囊菌门毛壳菌科比例明显提高[31],本研究结果与之一致。毛壳菌对病原菌的拮抗作用主要体现在重寄生和产生毛壳素等抗生素方面,是重要的植物病害生防真菌[32]。可见,施用微生物肥料有助于控制有机农田作物病害的发生。
土壤微生物类群复杂,微生物群落特征是微生物种群协同作用的结果,不能依照单一或某几个种群的变化阐释微生物肥料对有机农田土壤微生物群落的影响,后续研究将进行长期定位连续观测,结合土传病害严重度进一步揭示其影响作用。本研究发现,根区土壤微生物中未分类或未确定种属的物种也受微生物肥料的影响,受限于数据库的原因,未能解释,有待通过深度测序或利用其他先进手段对这些微生物进行更细致、更深入的分类研究。
参考文献:
[1]中华人民共和国国家质量监督检验检疫总局,中国国家标准化管理委员会.农用微生物菌剂:GB 20287—2006[S]. 北京:中国标准出版社,2006.
[2]许景钢,孙 涛,李 嵩. 我国微生物肥料的研发及其在农业生产中的应用[J]. 作物杂志,2016(1):1-6.
[3]国家认证认可监督管理委员会.中国有机产品认证与有机产业发展(2016)[M]. 北京:中国质检出版社,2017:1-3.
[4]Bokulich N A,Mills D A. Improved selection of internal transcribed spacer-specific primers enables quantitative,ultra-high-throughput profiling of fungal communities[J]. Applied and Environmental Microbiology,2013,79(8):2519-2526.
[5]Caporaso J G,Lauber C L,Walters W A,et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms[J]. The ISME Journal,2012,6(8):1621-1624.
[6]樓 骏,柳 勇,李 延. 高通量测序技术在土壤微生物多样性研究中的研究进展[J]. 中国农学通报,2014,30(15):256-260.
[7]鲁如坤. 土壤农业化学分析方法[M]. 北京:中国农业科学技术出版社,2000:107-109.
[8]Xu N,Tan G C,Wang H Y,et al. Effect of biochar additions to soil on nitrogen leaching,microbial biomass and bacterial community structure[J]. European Journal of Soil Biology,2016,74:1-8.
[9]Magoc T,Salzberg S L. FLASH:fast length adjustment of short reads to improve genome assemblies[J]. Bioinformatics,2011,27(21):2957-2963.
[10]Bolger A M,Lohse M,Usadel B. Trimmomatic:a flexible trimmer for Illumina sequence data[J]. Bioinformatics,2014,30(15):2114-2120.
[11]Quast C,Pruesse E,Yilmaz P,et al. The SILVA ribosomal RNA gene database project:improved data processing and web-based tools[J]. Nucleic Acids Research,2013,41(D1):D590-D596.
[12]Wang Q,Garrity G M,Tiedje J M,et al. Naive bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy[J]. Applied and Environmental Microbiology,2007,73(16):5261-5267.
[13]Pitta D W,Parmar N,Patel A K,et al. Bacterial diversity dynamics associated with different diets and different primer pairs in the rumen of Kankrej cattle[J]. PLoS One,2014,9(11):e111710.
[14]何 芝,赵天涛,邢志林,等. 典型生活垃圾填埋场覆盖土微生物群落分析[J]. 中国环境科学,2015,35(12):3744-3753.
[15]Chen H,Boutros P C. VennDiagram:a package for the generation of highly-customizable Venn and Euler diagrams in R[J]. BMC Bioinformatics,2011,12:35.
[16]杜思瑶,于 淼,刘芳华,等. 设施种植模式对土壤细菌多样性及群落结构的影响[J]. 中国生态农业学报,2017,25(11):1615-1625.
[17]Zhao S,Liu D Y,Ling N,et al. Bio-organic fertilizer application significantly reduces the Fusarium oxysporum population and alters the composition of fungi communities of watermelon Fusarium wilt rhizosphere soil[J]. Biology and Fertility of Soils,2014,50(5):765-774.
[18]杨亚东,王志敏,曾昭海. 长期施肥和灌溉对土壤细菌数量、多样性和群落结构的影响[J]. 中国农业科学,2018,51(2):290-301.
[19]Liu J J,Sui Y E,Yu Z H,et al. High throughput sequencing analysis of biogeographical distribution of bacterial communities in the black soils of northeast China[J]. Soil Biology and Biochemistry,2014,70:113-122.
[20]陈丹梅,段玉琪,杨宇虹,等. 长期施肥对植烟土壤养分及微生物群落结构的影响[J]. 中国农业科学,2014,47(17):3424-3433.
[21]Caporaso J G,Kuczynski J,Stombaugh J,et al. QIIME allows analysis of high-throughput community sequencing data[J]. Nature Methods,2010,7(5):335-336.
[22]吴江利,罗学刚,李宝强,等. 微生物菌肥作用下荒漠土壤微生物群落结构和功能研究[J]. 中国农学通报,2015,31(9):216-223.
[23]游 偲,张立猛,计思贵,等. 枯草芽孢杆菌菌剂对烟草根际土壤细菌群落的影响[J]. 应用生态学报,2014,25(11):3323-3330.
[24]康 捷,章淑艷,韩 韬,等. 两种麻山药典型病害根际土壤微生物多样性的研究[J]. 生物技术通报,2017,33(7):107-113.
[25]Luo J,Ran W,Hu J,et al. Application of Bio-Organic fertilizer significantly affected fungal diversity of soils[J]. Soil Science Society of America Journal,2010,74(6):2039-2048.
[26]Dawar S,Wahab S,Tariq M,et al. Application of Bacillus species in the control of root rot diseases of crop plants[J]. Archives of Phytopathology and Plant Protection,2010,43(4):412-418.
[27]Jiang Z Q,Guo Y H,Li S M,et al. Evaluation of biocontrol efficiencies of different Bacillus preparations and different field application methods against Phytophthora bight of bell pepper[J]. Biological Control,2006,36(2):216-223.
[28]彭 震,王春娟,陈庆河,等. 生物肥料“宁盾”对大豆疫霉病的防效及对毛豆的促生作用[J]. 上海农业学报,2014,30(6):95-98.
[29]韩 哲,刘守伟,潘 凯,等. 不同栽培模式对黄瓜根际土壤酶活性及细菌群落结构的影响[J]. 植物营养与肥料学报,2012,18(4):922-931.
[30]姜 飞,刘业霞,艾希珍,等. 嫁接辣椒根际土壤微生物及酶活性与根腐病抗性的关系[J]. 中国农业科学,2010,43(16):3367-3374.
[31]李发虎,李 明,刘金泉,等. 生物炭对温室黄瓜根际土壤真菌丰度和根系生长的影响[J]. 农业机械学报,2017,48(4):265-270,341.
[32]Di Pietro A,Gut-Rella M,Pachlatko J P,et al. Role of antibiotics produced by Chaetomium globosum in biocontrol of Pythium ultimum,a causal agent of damping-off[J]. Phytopathology,1992,82(2):131-135.
