
Mn掺杂InP(111)-In极化面电子结构与磁性的第一性原理研究
2019-09-18 02:52付斯年郑友进
人工晶体学报 2019年8期
付斯年,李 聪,郑友进
(牡丹江师范学院物理系,黑龙江省新型碳基功能与超硬材料重点实验室,牡丹江 157011)
1 引 言
近年来,稀磁半导体由于其兼具电荷与自旋的特性而被人们广泛关注[1]。其中,Ⅲ-Ⅴ族化合物InP表现出了卓越的物理性能[2-4]。尤其是Mn掺杂InP在光电器件应用领域显示出了巨大的应用潜力。Khalid等[5]证明了通过脉冲激光退火可提高Mn掺杂InP的磁学性能。Yoon等[6]报告了在较低的退火温度条件下可制备InMnP∶Zn外延层。Khalid等[7]证明了在低温环境下,Mn掺杂InP可展现出阶跃电导。近些年来,InP基稀磁半导体迅速成为了热门研究领域[8]。
与体位结构相比,表面原子由于其独特的物理化学性质而引起人们广泛关注[9],这主要源于表面原子含有大量不饱和的悬空键[10],因此,研究Mn掺杂InP体系的表面态具有重要意义。另外,Ⅲ-Ⅴ族化合物稀磁半导体的铁磁性起源一直保持争议,一方面认为稀磁半导体磁性源于价带中的p-d轨道交换作用,该理论可通过Zener的平均场理论解释[11]。另一方面认为铁磁性主要源于Mn原子受主能级的位置[12],然而,关于Mn掺杂InP表面态的理论鲜有报道,本文通过模拟计算研究了Mn掺杂InP(111)-In极化面的电子结构与磁学特性。得出的结果可为进一步研究InP基稀磁半导体做理论参考。
2 理论模型与计算方法
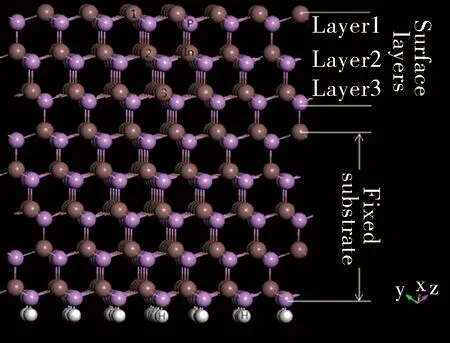
图1 InP (111)方向表面模型Fig.1 InP (111) surface model
本文基于密度泛函下的平面波超软赝势法,并通过广义梯度近似描述交互关联能。所有的计算工作均通过CASTEP计算模块完成[13]。价电子组态分别为In-4d105s25p1、 P-3s23p3与Mn-3d54s2。能量计算收敛精度为1.0×10-6eV/atom。平面波截断能为400 eV。本文采用GGA+U的方法修正由广义梯度近似(GGA)引起的带隙误差。如表1所示,对于纯净In128P128体系而言,当UIn-s值选取为5.00 eV,UP-p值选取为6.00 eV时,计算得到带隙宽度为1.351eV,这与本征InP禁带宽度(Eg=1.344 eV)相符。UMn-d值则选取为4 eV[14-15]。所有关于电子态密度计算均采用自旋极化处理。表面优化后的结构如图1所示,InP表面模型由8个(111)方向的In-P原子层组成。在几何优化的过程中,最上面3层采用弛豫处理,底下5层作为基底用来模拟大块晶体InP。如表1所示,经优化后得到的晶格常数的计算值与理论值之间符合较好, 相对误差不超过0.4%,这说明本文计算方法合理。为了稳定P终极面电荷,本文采用赝H钝化了P终极面[16],其中赝H原子的分数电荷选取为0.75e-[17-18]。同时,在(111)方向创建了1.5 nm厚的真空层。所有的Mn掺杂InP表面模型建立如下:纯净In128P128表面、In127Mn1P128、 In127Mn2P128及 In127Mn3P128。Mn原子符号的上角标代表Mn原子替换In原子的位置,如图1所示。

表1 取不同U值时掺杂体系的禁带宽度与晶格常数Table 1 The band gap and lattice constant of doping system with different U values
3 结果与讨论
3.1 掺杂体系形成能分析
为了确定掺杂对于体系结构稳定性的影响,所有的Mn掺杂表面模型的形成能计算如下[19]:
Ef=Edoping-Epure+EIn-EMn
其中Edoping是Mn掺杂InP体系的基态能量,Epure是纯净InP表面体系的基态能量,EIn与EMn分别是In原子与Mn原子的化学势。
如表2所示,随着Mn原子的掺杂位置上移,形成能Ef逐渐降低。这说明Mn处在最表面层时体系的形成能最低,体系最稳定。这一结论与文献[20]的实验结果相一致。表2显示出Mn在不同掺杂位置时所对应的最长键长与最短键长。如表2所示,随着Mn原子掺杂位置上移,Mn-P最长键与最短键的键长差异增大(对于位置1的Mn原子是由于出现了悬空键,悬空键键长为0.1603 nm,如图1)。这说明掺杂位置越靠近表面,晶格畸变越严重。这种晶格畸变使Mn原子的掺杂变得更加容易,因此所需要的形成能更低。这与之前对体系形成能的分析结果相一致。由于Mn的离子半径(0.067 nm)略小于In的离子半径(0.080 nm)。因此,Mn掺杂InP(111)-In极化表面相对稳定。

表2 所有体系的形成能、Mn-P键长、净磁矩与铁磁-反铁磁态能量差ΔETable 2 Formation energy, Mn-P bond length, magnetic moment and energy difference ΔE of all models
3.2 态密度分析
本文计算了纯净InP表面体系与所有Mn掺杂InP表面体系的自旋态密度。如图2所示。
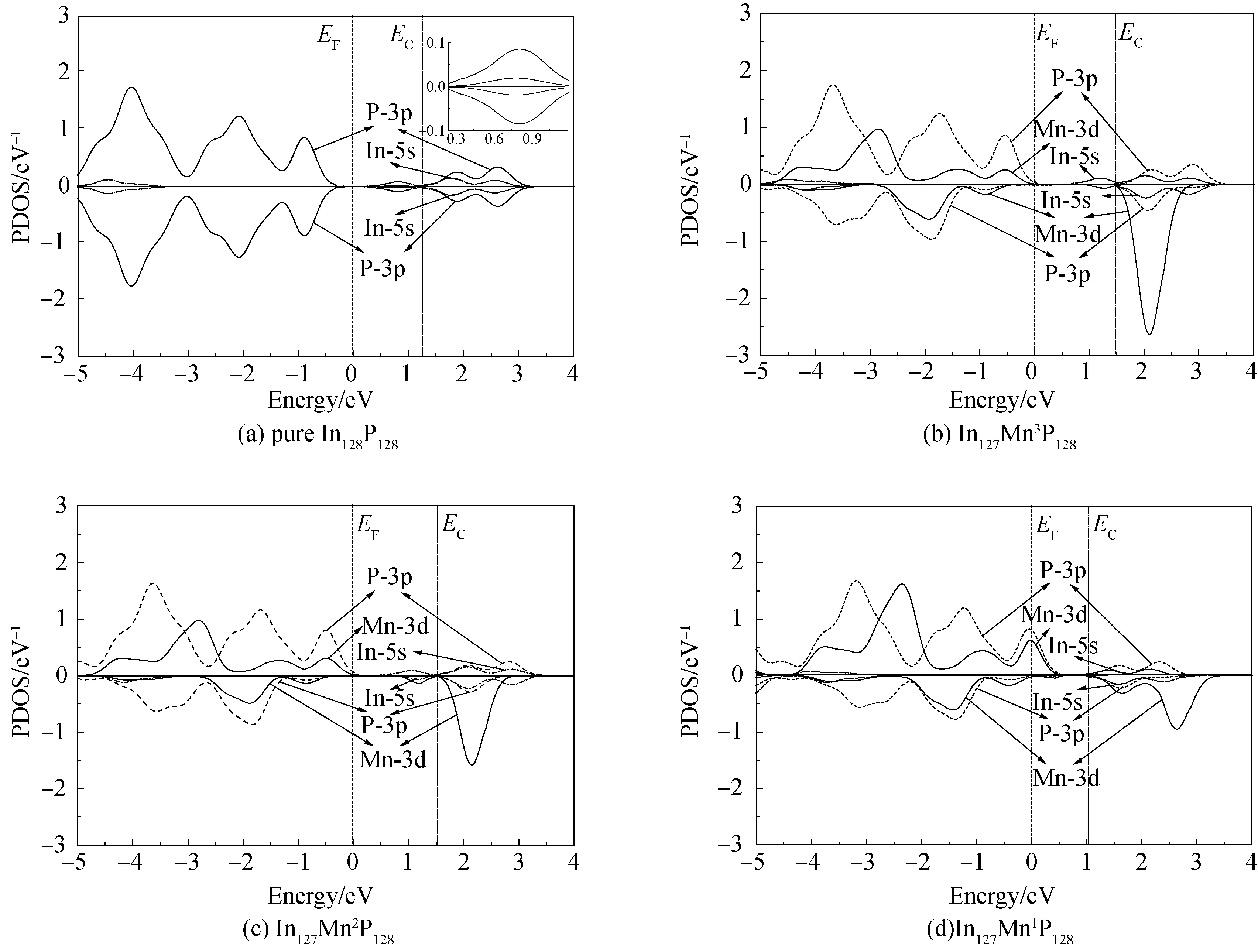
图2 所有模型的自旋态密度分布Fig.2 SDOS of the models
如图2(a)所示,在纯净InP (111)-In极化面体系中,费米能级EF位于带隙且有若干表面能级存在于带隙中。这些表面能级主要由In-5s态及少量P-3p态所贡献。并且在纯净体系中可以看出,体系的电子自旋态密度具有对称性,整个体系并未显示出铁磁性特征。然而在表2中可以看出,在所有的Mn掺杂InP表面体系中,掺杂体系的铁磁-反铁磁能量差ΔE都是负值,这说明在所有掺杂体系中铁磁基态是稳定态。如图2(b),2(c)和2(d)所示,掺杂体系的电子自旋态密度都具有不对称性,并展现出了明显铁磁性特征。
如图2(b)所示,在体系In127Mn3P128中,费米能级向低能级方向移动并靠近价带顶。整个体系呈现出一定P型半导体特征。具有不对称性的Mn-3d自旋态密度主要分布在费米能级附近的1.5~2.8 eV能量范围区。在Mn-3d态的诱导极化作用下,P-3p态产生了一定程度的不对称的自旋电子态密度,因此整个体系呈现出铁磁性特征。经计算,其净磁矩为4.03 μB。在体系In127Mn3P128中替换In原子的Mn原子提供3个电子与3个P-3p态电子成键。因此Mn原子的氧化态是+3[21]。根据原子物理可知,一个自旋电子贡献1 μB的自旋磁矩,大约相当于4.03 μB的四分之一。因此,体系In127Mn3P128的净磁矩主要由4个未成对的Mn-3d电子所贡献。从图2(b)可知,在体系In127Mn3P128中,若干表面态能级出现在导带底。与纯净体系相比,体系In127Mn3P128呈现出一定程度的半金属性。如图2(b)所示,该体系的价带顶主要由P-3p态与Mn-3d态构成。并且与纯净体系相比,In127Mn3P128体系的费米能级更接近价带顶,整个掺杂体系表现出P型半导体特征。导带底的表面态能级主要由In-5s电子态所贡献。由表2可知,在体系In127Mn3P128中,Mn-P最长键键长与最短键键长相差0.002 nm。这说明Mn原子与最邻近4个P原子所形成的正4面体结构保持比较完好,依然保持着较好的空间对称结构。
与图2(b)相比,由图2(c)可以看出,体系In127Mn2P128在价带顶费米能级附近的p-d轨道杂化程度增强。并且与体系In127Mn3P128相比,In127Mn2P128体系的费米能级向低能级方向有微小移动,掺杂体系的p型半导体特征有所增强。如图2(c)所示,与In127Mn3P128相比,In127Mn2P128体系中的Mn原子的自旋态密度在高能级区域呈现出一定程度的弱局域特征。并且该体系中Mn-P最长键键长与最短键键长相差0.0046 nm,相对差距为2%。这说明Mn原子周围的正四面体的空间对称结构遭到较小破坏,如表2所示,此时体系的净磁矩为4.07 μB,Mn原子的氧化态介于+2与+3之间,后文将证实这一观点。
由图2(d)可以看出,在In127Mn1P128体系中,Mn原子的自旋态密度在高能级区域的弱局域特征明显增强,自旋态密度曲线变得弥散。如图2d所示,与In127Mn2P128相比, In127Mn1P128体系中的p-d轨道在费米能级附近的杂化程度进一步增强。与此同时,体系的费米能级向低能方向移动明显并进入了价带顶。整个体系呈现出明显的P型半导体特征。如图2d所示,掺杂体系的表面态能级主要由Mn-3d,In-5s 以及 P-3p 态构成。如表2所示,In127Mn1P128体系的净磁矩为5.06 μB。大约相当于5个Mn-3d自旋电子的贡献。因此,在In127Mn1P128中Mn原子的氧化态为Mn2+。
3.3 氧化态转变分析
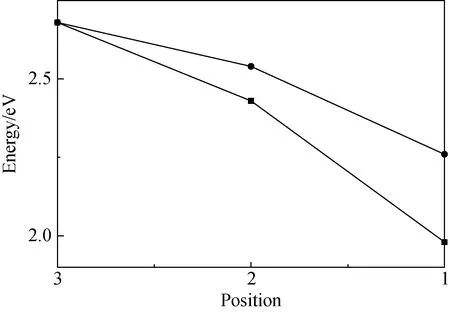
图3 不同体系中体系的形成能随Mn原子不同 掺杂位置的关系Fig.3 The formation energy of Mn atom at different position in diferrent systems
为了进一步证实随着掺杂位置上移,Mn原子氧化态由Mn3+转化为Mn2+这一结论,本文做了如下计算。首先假设在各个不同掺杂位置Mn原子的氧化态都是Mn3+,之后再分别计算各个不同掺杂位置的体系的形成能。如图3所示,图中圆点表示各个假设体系的形成能随Mn原子掺杂位置变化关系。方块表示Mn原子氧化态转变的体系(本文研究的体系)的形成能随Mn原子掺杂位置的变化关系。在图3中可以看出,如果Mn原子的氧化态不随着掺杂位置上移而改变,那么该体系的形成能就会与Mn原子氧化态转变的体系的形成能差距越来越大。这说明Mn原子氧化态不变的体系不能稳定存在。这直接证明了随着掺杂位置上移,Mn3+逐渐转化为Mn2+这一结论的正确性。
另外,从相邻原子层之间形成的电偶极矩的角度也可对Mn原子氧化态转变作出分析。所有的表面掺杂模型都由沿(111)方向的重复的P-In原子层所组成。P原子层与In原子层带有相反电量。因此在(111)方向形成了电偶极矩,为了消除由电偶极矩而产生的内建电场的影响,如前文所述,在P终极面采用赝H进行钝化,从而使P终极面负电荷减少[22]。但在In终极面并没有采用电负性强的原子(如F原子)进行钝化。因此会在In终极面集居一部分负电荷。在这种条件下,由于Mn3+本身具有较强的氧化性,因此不可能稳定存在,Mn3+离子会俘获一部分电子从而由Mn3+转化为Mn2+,在这一过程当中,Mn3+离子充当了一个受主离子的角色。这也是随着Mn原子掺杂位置上移,掺杂体系的P型半导体特征明显增强的原因,这一结论与之前的态密度的结果相一致。
下面进一步分析Mn原子周围的空间对称结构的改变对Mn原子氧化态转变的影响。从表2可以看出,与In127Mn3P128体系相比,In127Mn1P128体系中的Mn-P键键长明显变短,最短键长为为0.2296 nm。产生这一现象的主要原因是,处在表面层的Mn原子在(111)方向形成了悬空键,悬空键的存在导致Mn原子只能与剩余的3个P原子成键,Mn原子悬空键导致Mn原子与最邻近的P原子所形成的正四面体的空间对称性消失。Mn-P键键长变短导致Mn-P原子之间p-d轨道杂化程度加剧,这一结论与前文态密度分析相一致。p-d轨道杂化程度加剧使Mn原子有更大几率从P原子俘获电子,从而完成Mn3+到Mn2+氧化态的转变。
4 结 论
本文利用密度泛函的广义梯度近似研究了Mn掺杂InP(111)-In极化面的电子结构与磁学性质,得出以下结论:
(1)随着Mn原子掺杂位置靠近In极化面,Mn原子掺杂的形成能逐渐降低,并且所有Mn掺杂表面模型均表现出稀磁半导体特征,这是由于费米能级附近的Mn-3d自旋态密度的非对称性所导致的。
(2)通过对掺杂体系形成能与净磁矩的分析发现,所有掺杂在表面层的Mn原子的氧化态都是Mn2+。
(3)随着Mn掺杂位置上移,掺杂体系的费米能级向低能级方向移动并进入价带顶,表面体系表现出明显的的P型半导体特征。
猜你喜欢
当代党员(2022年9期)2022-05-20
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
中外文摘(2021年7期)2021-04-23
华人时刊(2021年23期)2021-03-08
发明与创新·小学生(2020年10期)2020-10-19
复旦学报(医学版)(2020年3期)2020-06-18
发明与创新·小学生(2019年12期)2019-12-05
中学生数理化·高三版(2017年1期)2017-04-20
