
吩嗪衍生物晶体结构预测及其动力学研究
2019-10-15 07:38薛君
山东化工 2019年18期
薛 君
(中国海洋大学 化学化工学院,山东 青岛 266510)
晶体在不同的体系中会生长成不同的几何构型,在不受抑制体系中自发地长成对称多面体,而在受抑制的生长体系中生长时,就会具有特殊的外形[1]。现阶段的晶体预测主要采用MD、kMC和连续模拟的组合,使用多尺度整合方法对晶体结构进行探究,但是这种方法没有考虑溶剂的影响并且只适用于部分体系[3]。分子动力学(MD)模拟提供了一种研究分子晶体生长的有效方法,因为它们不仅限于单一结构,而是可以计算集合构象的性质,因此完全结合了熵的贡献[4]。使用MD模拟作为基础,允许探索更大的尺寸和更长的时间长度[5]。
本文将有机小分子体系从分子自组装行为开始进行动力学模拟,探究晶体生长过程,并根据分子自组装技术聚集形态进行晶体结构模拟,进而探究分子自组装技术与晶体结构形成的机理。以2-苯基-1H-咪唑并[4,5-b]吩嗪(PND)为例[6],对其进行量子化学计算和动力学模拟计算,将其放入搭建好的溶剂环境中,模拟出最稳定的结构进行晶体预测,并和实际晶体结构进行对比验证,进而总结出进行晶体预测的实验方法,有效对有机小分子自组装晶体结构进行预测。
1 实验部分
1.1 量子化学计算
搭建吩嗪分子结构,由M06-2X / cc-PVDZ基组对PND分子进行优化和能量计算,得到几何结构的特征参数,如能量,偶极矩,HOMO和LUMO的电荷密度以分析分子的反应活性位点,所有计算均在Gaussian09软件中进行。
1.2 分子动力学模拟
由高斯优化过的分子结构引入MS软件中使用Focite模块对PND进行结构优化和MD模拟,分子构型优化选择COMPASS力场,步数设置为500000步,使其达到能量最低的平衡状态。动力学模拟也是在COMPASS力场下进行的,选择 NPT系综,模拟开始于积分步骤1fs,温度设置为298K,压力P = 1atm(常温常压条件)。
1.3 溶剂中的分子动力学模拟
DMF溶剂盒子的密度设置为0.908,根据PND的分子量以及分子体积确定溶剂盒子的分子个数为1024,确保其不会产生分子离域现象,溶剂盒子c方向上的长度为6nm,将PND小分子放入构建好的溶剂盒子中进行结构优化和分子动力学计算,选用Focite模块中的COMPASS力场,NPT(恒温恒压)系综,模拟开始于积分步骤1fs,步数设为500000步,温度T = 298K(恒温器设为Nose Themostat),压力P = 1atm。探究PND分子在溶剂中的自组装行为。
1.4 晶体结构预测
将DMF溶剂中的PND所得结果创建运动组,对PND进行晶体结构预测。使用MS中的Polymorph模块,选择COMPASS力场,对PND1的S1,S2结构进行Prediction计算,选择的空间群为P21/C,P-1,P212121, C2/C, P21和PBCA。
2 结果与讨论
2.1 量子化学计算
由前线轨道理论可知,HOMO轨道的能级越高与失去电子的能力成正比;LUMO的能级与分子得到电子的能力成反比。图1为PND分子的HOMO,LUMO 轨道图,结合表1分子特征原子对轨道的贡献,可以分析出分子的反应活性位点。
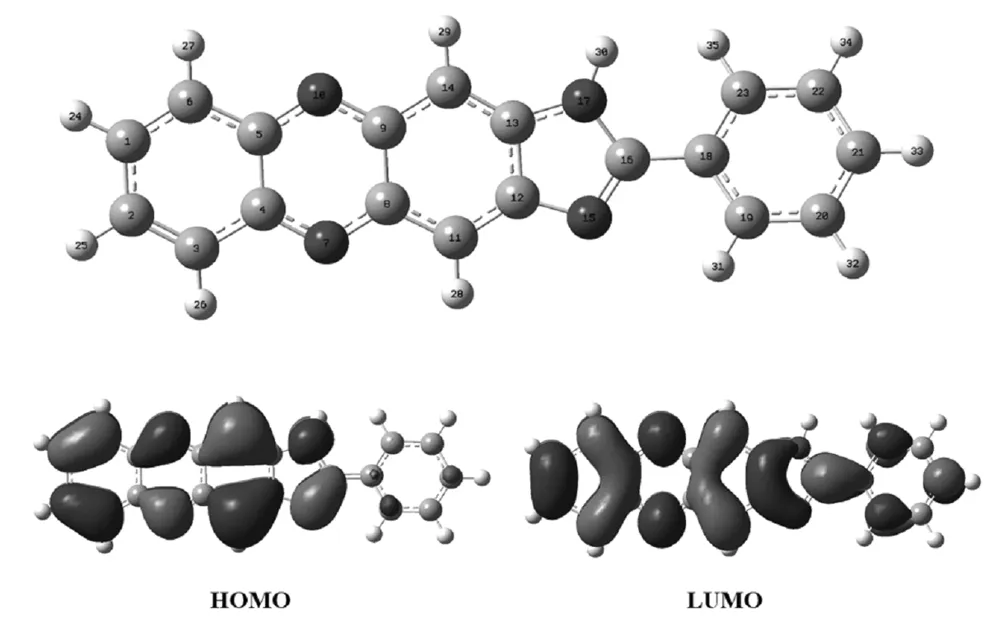
图1 PND分子的前线轨道分布图
由图1和表1可以看出,PND分子中最高占据轨道(HOMO)主要分布在N7、N10、N15和N17原子上,其中N7对HOMO贡献最大达到5.26%,其余部分分布在 N10和N15原子上,但是贡献率数值相差不大;最低空轨道主要分布在N7原子上,N10原子上也占有一定分布,但是N15和N17原子对LUMO轨道的贡献相比较下就显得很小了。结合表1中各个原子所带的电荷可推测,PND分子中的氮原子都带有负电荷,亲核性能较强,其中N7和N10原子应该为亲核活性中心,由于PND中带有活泼氢H30,所以极易形成N-H…N键,这是PND分子自组装结构形成的主要作用力机制。
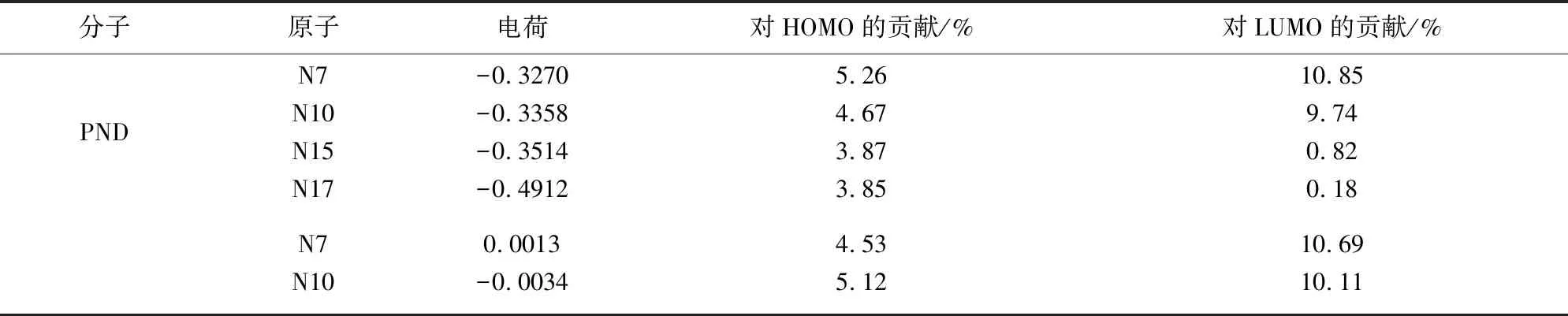
表1 PND分子中N、O原子电荷分布和对前线轨道的贡献
2.2 分子动力学模拟
由量子化学计算结果可知,N17上有一个裸露的原子氢,所以极易与其分子内的N7、N10以及N15形成N-H…N键,但是单从这几个氮原子对HOMO、LUMO轨道的贡献上不能确定H30会与哪一个氮原子成键,所以我们在MS软件中构建分子结构时对分子可能产生的自组装结构进行合理的猜想,并从空间以及成键上设计出了以下5种情况。
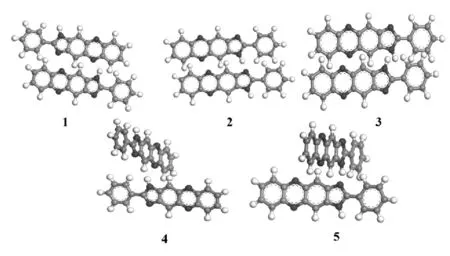
图2 PND分子的自组装假想模型
将这些假象模型在COMPASS立场下对分子进行优化后开始动力学计算,得到五种结构趋于最优化的结果。结果显示不同的假想模型在计算后在空间中趋于相似的层状结构。

表2 五种假想模型的能量变化
由表2中可以看出,五种假象模型的初始能量有一定的差异,但经过分子优化和动力学计算之后最终能量相差不大,说明形成的最终结构相似。逐个增加分子个数并对可能产生的自组装结构进行猜想,然后用同样的方法对结构能量最低化后进行动力学模拟计算观察结构的空间构型变化以及键长键角等参数的变化,确定分子自组装稳定结构。
由图3可知,PND分子在真空中的自组装趋于空间层状重叠。从图2中可以看出,初始放置时两个PND分子存在一定的夹角并且氢键组合方式也不相同。当体系达到平衡后,PND分子发生扭转并且氢键键长和键角也发生了明显变化。分子自组装结构中的N17-H30…N7与N17-H30…N15氢键键角都趋向于180°,并且键长长度也有不同程度的增加,但仍在正常范围内,这也说明PND分子在真空中确实可以形成稳定的分子自组装结构。
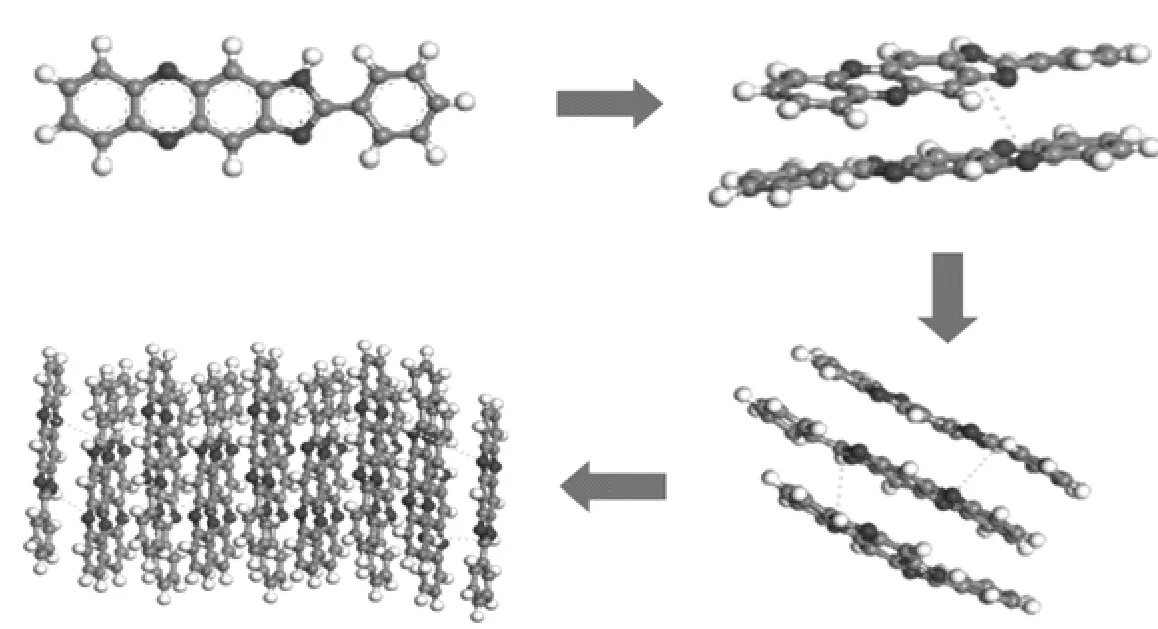
图3 PND分子在真空中的自组装结构
2.3 溶剂中的分子动力学模拟
由2.2结果可知,PND分子形成的N17-H30…N7氢键和N17-H30…N15形成的氢键强度相差不大,所以将两个假想构型S1、S2放置于DMF溶剂盒子中进行多次动力学模拟,S1是两个PND分子通过N17-H30…N7氢键相连,再通过N17-H30…N15氢键连接另一个分子,S2是三个PND分子通过N17-H30…N15氢键形成的自组装结构。
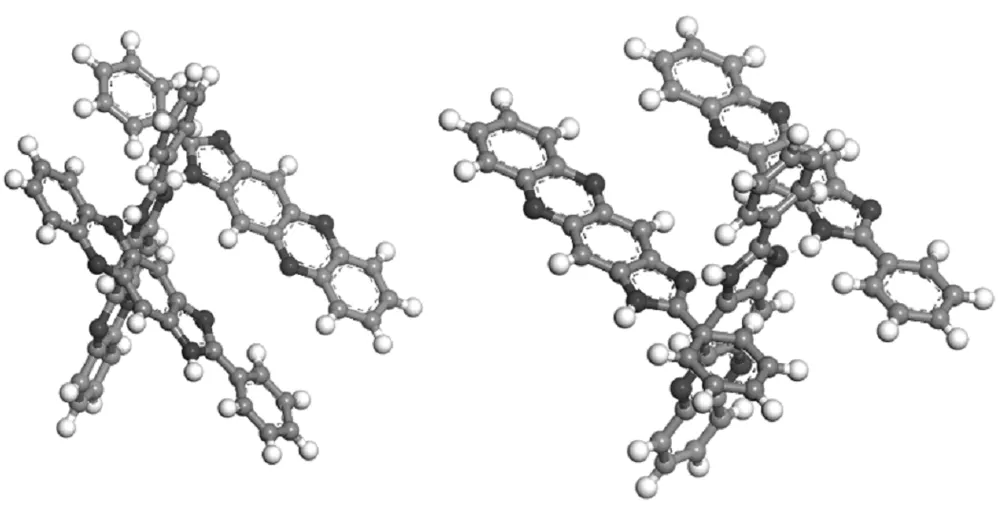
图4 PND分子的S1、S2结构图
由图5可以看出,PND分子在溶剂环境中的排列方式与在真空中不同。S1结构在DMF溶液中空间结构发生了较大的变化,π-核心与吩嗪基团反平行排列,吩嗪基团几乎平行于π-表面,而另一侧与π-表面相对应,与实验所观察到的结构基本相符,这就为我们在下一节的晶体结构预测中提供了动力组创建的依据。S2结构在DMF溶液中的空间构型趋近于真空中的结构,但是其中一个氢键N17-H30…N15键长为3.615Å,属于弱氢键作用。说明溶剂环境中的自组装行为可能较真空环境差异较大,真空环境下的自组装结构只能为实际自组装行为提供结构参考。

图5 PND在DMF溶剂中的自组装结构
2.4 晶体结构预测
空间群是晶胞中全部对称要素的组合。不同空间群晶体结构如图6所示。P21-C、C2-2和P2空间群都属于单斜晶系,P212121和PBCA空间群属于正交晶系,而P-1空间群属于三斜晶系[7]。表3中显示了各个空间群的占比,总能量以及电荷等参数,从表中可以看出,虽然PBCA空间群形成的晶体占比较小,但是其晶体结构的能量最小,说明其形成的结构最为稳定。实验所得晶体为正交晶系,结构初步吻合。

图6 模拟所得不同空间群的晶体结构
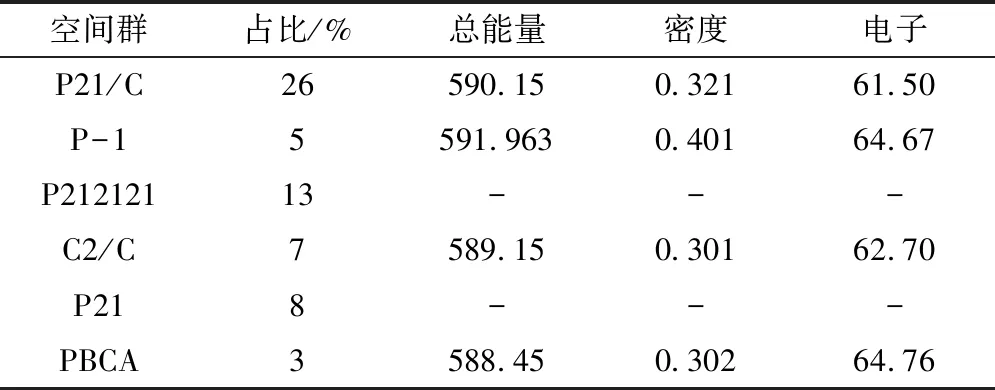
表3 模拟晶体各个空间群计算所得参数
将PBCA空间群形成的晶体结构以及在CCDC结构库里查找到的PND晶体结构在MS中,在其达到稳定平衡状态后进行CASTEP计算,得到晶体结构中氢键键长和粒子分布如表4和表5,分别是实验晶体和模拟晶体的计算结果。分析数据可知,两种晶体形成分子间氢键的位置大相同,模拟晶体中的分子间氢键长度比实验晶体中略大一些,但数值相差不大。
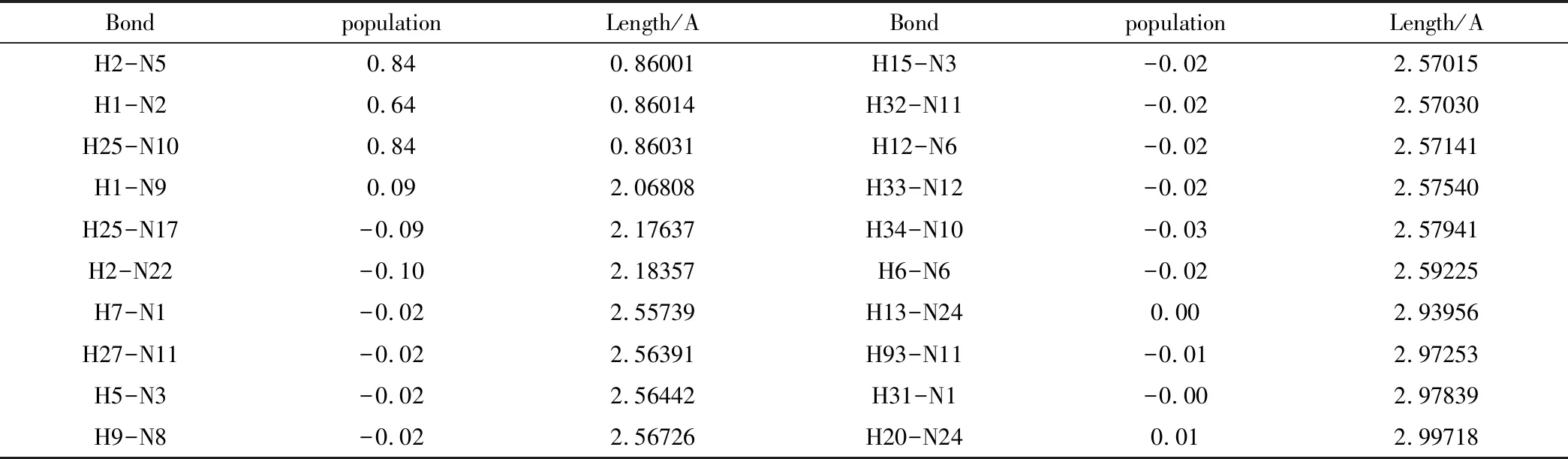
表4 实验晶体氢键键长和粒子数
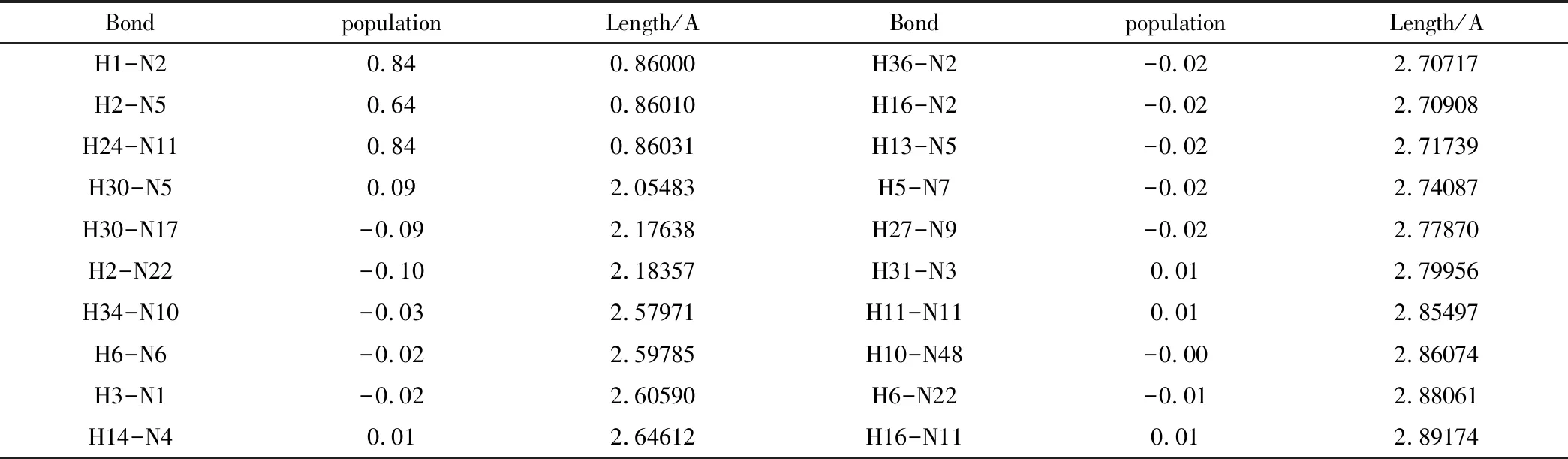
表5 模拟晶体氢键键长和粒子数
图7中,上图为在CCDC库中引入的PND晶体结构在MS中计算所得射线粉末衍射图,下图为预测PBCA空间群下的晶体结构计算所得射线粉末衍射图,两图进行比较可以看出其特征峰位置基本重合,可以推测出两者属于同一种晶体,从而证明PND分子的晶体结构预测方法是可行的。
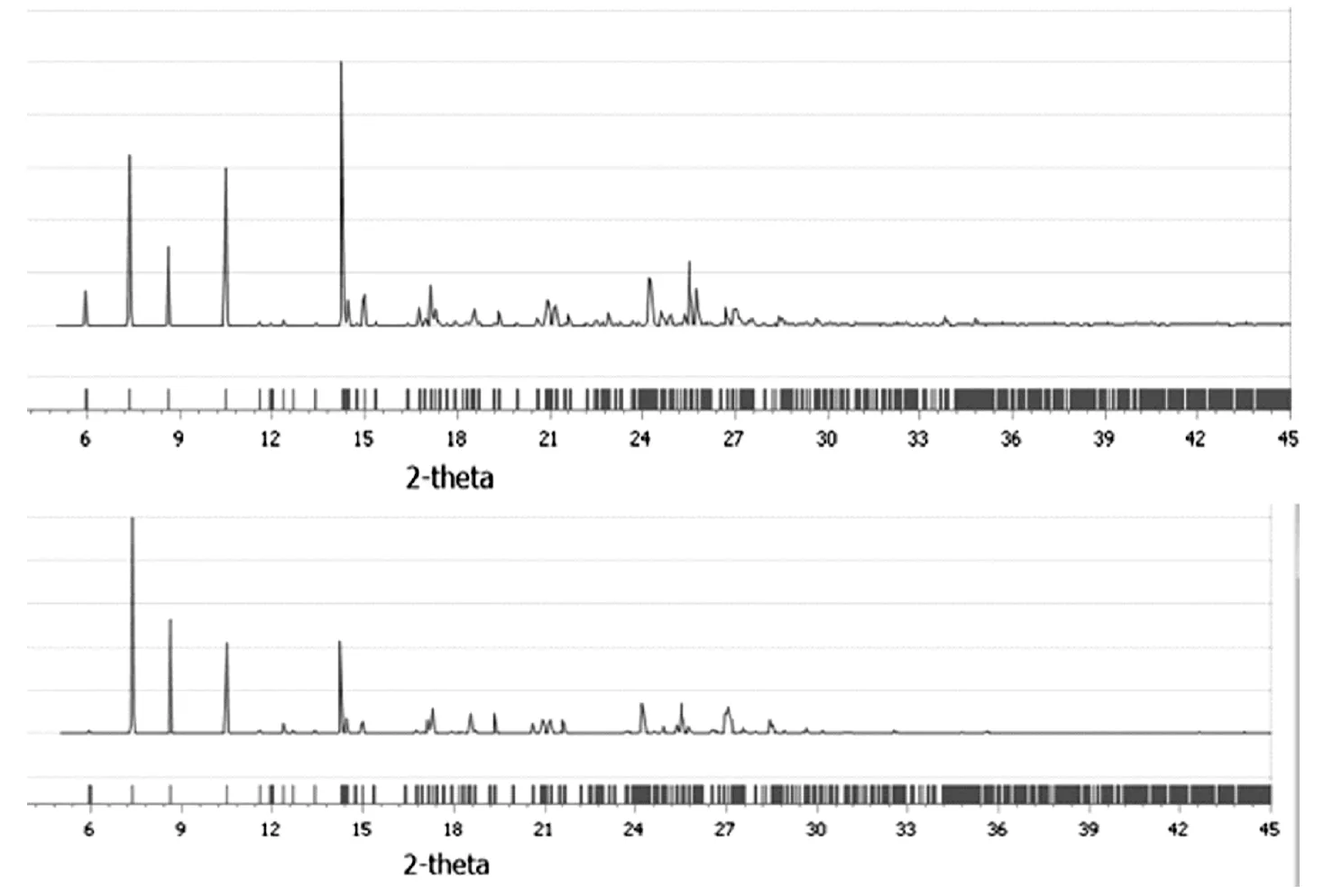
图7 CCDC数据库中PND1晶体理论与预测X-射线粉末衍射图
3 结论
利用DFT/MD方法对2-苯基-1H-咪唑并[4,5-b]吩嗪分子进行自组装模拟和晶体结构预测,从分子层面研究了晶体生长机理。模拟所得结果和实验结果具有良好的一致性,说明由分子自组装行为进而推测分子晶体结构的预测方法是可行的,为类似氢键驱动的分子自组装体系小分子晶体的结构预测提供了一种新的方法。
猜你喜欢
人工晶体学报(2021年10期)2021-11-26
波谱学杂志(2021年3期)2021-09-07
——以高中化学“氢键”的教学为例
教学月刊(中学版)(2020年13期)2020-12-29
中学生数理化(高中版.高考数学)(2020年2期)2020-04-21
中学生数理化(高中版.高考理化)(2020年2期)2020-04-21
天津师范大学学报(自然科学版)(2016年4期)2016-12-14
中学化学(2015年12期)2016-01-19
火炸药学报(2014年3期)2014-03-20
郑州大学学报(理学版)(2013年2期)2013-03-11
