
2 种斯皮诺素固体分散体的制备及其体内药动学行为
2019-10-16 12:47张铁山尚曙玉王聪颖张智强
中成药 2019年9期
张铁山, 尚曙玉, 王聪颖, 张智强
(1.黄河科技学院,河南 郑州450005;2.天津药物研究院药业有限责任公司,天津300301)
斯皮诺素是从鼠李科植物酸枣Ziziphus jujube Mill.var.spinosa(Bunge)Hu ex H.F.Chou 中分离得到的黄酮类化合物[1],其性平,味甘、酸,归肝、胆经,具有抗抑郁、抗心律失常、催眠、镇静、抗氧化等多种药理活性[1-3],但其水溶性很差[4],从而限制了疗效发挥及临床应用。作为药食同源两用品[5],开发出斯皮诺素高生物利用度制剂具有重要的现实意义,但目前国内外鲜见相关报道[6]。
固体分散体[7-9]由于制备工艺相对简单,常作为改善中药难溶性药物的制剂技术之一,而近年来磷脂复合物固体分散体也逐渐受到国内外专家学者的关注[10-12],它是在一定条件下将难溶性药物与磷脂制成磷脂复合物后,进一步采用亲水性高分子材料形成固体分散体。本实验分别制备了斯皮诺素固体分散体、磷脂复合物固体分散体,并比较了两者体内药动学行为。
1 材料
1.1 仪器 Agilent 1260 型高效液相色谱仪(美国Agilent 公司);Bell-ennium 型加热磁力搅拌器(上海书俊仪器有限公司);FA1004 型电子分析天平(上海光学仪器厂);XD-5000A 型真空旋转蒸发仪(上海贤德实验仪器有限公司);XW-80A 型漩涡混合仪(上海医科大学仪器厂);TF-008-W 型氮气吹扫仪(瑞诚科学仪器公司);HTZ-92-01 型恒温振荡器(上海精怡科学仪器公司);D8 型粉末衍射仪(德国布鲁克公司)。
1.2 试药 斯皮诺素对照品(批号T171220,瑞芬思生物科技有限公司);斯皮诺素原料药(批号160615001S,湖北巨胜科技有限公司, 含有量>98%);盐酸普萘洛尔(批号100783-201401,中国食品药品检定研究院);卵磷脂(批号PC-98T,辅必成上海医药科技有限公司);聚乙烯吡咯烷酮(PVK K30, 批 号 25000240379, 亚 什 兰 集 团公司)。
1.3 动物 清洁级SD 大鼠,体质量200 ~240 g,购自河南省动物实验中心,许可证号SCXK(豫)2016-0001,实验前1 d 领取。
2 方法与结果
2.1 斯皮诺素含有量测定
2.1.1 色谱条件 Agilent Eclipse Plus C18色谱柱(250 mm×4.6 mm,5 μm);流动相乙腈-0.15%磷酸(40 ∶60);检测波长336 nm;柱温35 ℃;体积流量1.0 mL/min;进样量20 μL。
2.1.2 方法学考察 精密称取斯皮诺素对照品10.0 mg 至50 mL 量瓶中,乙腈超声溶解,定容至刻度,混匀,取适量用流动相逐步稀释,配制成50.0、 25.0、 5.0、 0.5、 0.05 μg/mL, 在“2.1.1” 项色谱条件下进样测定,以溶液质量浓度为横坐标(X),峰面积为纵坐标(Y) 进行回归,得方程为Y=1.041 2X-1.208 3(r=0.999 8),在0.05 ~50.0 μg/mL 范围内呈良好的线性关系。取低 (0.05 μg/mL)、 中 (25.0 μg/mL)、 高(50.0 μg/mL) 质量浓度对照品溶液,在“2.1.1”项色谱条件下进样测定,测得峰面积RSD 均小于0.45%,表明仪器精密度良好;平均加样回收率分别为99.01%、100.26%、100.09%,RSD 均小于1.66%;室温下于5 d 内在“2.1.1” 项色谱条件下进样测定,测得峰面积RSD 为0.66%,表明溶液在5 d 内稳定性良好。
2.2 固体分散体制备及X 射线粉末衍射(XRPD)分析 采用溶剂挥发法制备固体分散体。取斯皮诺素0.2 g、PVP K30 1.2 g,加入100 mL 无水乙醇中,50 ℃下搅拌4 h,减压旋蒸尽量除尽有机溶剂,即得,置于45 ℃真空干燥箱中24 h,敞口置于干燥器中。
XRPD 扫描分析条件为铜靶;管压40 kV;扫描范围3°~45°;扫描速度8°/min,结果见图1。由图可知,斯皮诺素以典型结晶状态存在,而在固体分散体中转变成无定型状态;同比例物理混合物中原料药的特征晶型峰仍可观察到,表明其结晶状态未得到改变。

图1 固体分散体XRPD 图Fig.1 XRPD pattern for solid dispersions
2.3 磷脂复合物固体分散体制备及XRPD 分析 取斯皮诺素0.2 g、卵磷脂0.25 g,置于50 mL 四氢呋喃中,45 ℃水浴搅拌4.0 h,真空旋蒸除去四氢呋喃,即得磷脂复合物。取0.2 g,按“2.2” 项下方法制备,即得。
对磷脂复合物及其固体分散体进行晶型分析,结果见图2。由图可知,斯皮诺素典型晶型峰在磷脂复合物中消失,呈无定型状态;制成磷脂复合物固体分散体后,它同样以无定型状态存在,表明制备成功。

图2 磷脂复合物固体分散体XRPD 图Fig.2 XRPD pattern for phospholipid complex solid dispersions
2.4 表观溶解度比较 取过量斯皮诺素、固体分散体、磷脂复合物、磷脂复合物固体分散体,加到10 mL 经超声处理后的蒸馏水中,固定于25 ℃恒温摇床上平衡2 d,15 000 r/min 下离心10 min,0.45 μm 微孔滤膜过滤,取续滤液,测定溶解度,结果见表1。由表可知,固体分散体、磷脂复合物、磷脂复合物固体分散体均可显著提高斯皮诺素溶解度(P<0.01)。
2.5 体外溶出度比较 取斯皮诺素、固体分散体、磷脂复合物、磷脂复合物固体分散体适量(以斯皮诺素计,含有量均为30 mg),2 mL 蒸馏水制成混悬液后置于活化处理的透析袋(7 000~14 000 Da)中,温度37 ℃,转速100 r/min,以900 mL 超声处理后的蒸馏水为释放介质,于180 min 内不同时间点取样3 mL,补加释放介质以保持总体积不变,0.45 μm 微孔滤膜过滤,HPLC 法测定斯皮诺素含有量,绘制溶出曲线,结果见图3。由图可知,固体分散体、磷脂复合物、磷脂复合物固体分散体可提高斯皮诺素体外溶出度,以固体分散体、磷脂复合物固体分散体更明显。
表1 溶解度考察结果(,n=3)Tab.1 Results of solubility investigation(,n=3)

表1 溶解度考察结果(,n=3)Tab.1 Results of solubility investigation(,n=3)
注:与斯皮诺素比较,**P<0.01
样品 1表观溶解度2/(μ g·mL-1)3平均值/(μg·mL-1)斯皮诺素 12.43 12.40 12.47 12.43±0.04固体分散体 139.70 139.92 139.86 139.82±0.11**磷脂复合物 26.53 26.39 26.44 26.45±0.07**磷脂复合物固体分散体 144.23 144.30 144.24 144.26±0.04**

图3 样品体外溶出曲线Fig.3 In vitro dissolution curves for samples
2.6 体内药动学行为研究
2.6.1 灌胃液制备 取斯皮诺素及其固体分散体、磷脂复合物固体分散体适量,0.5%CMC-Na 溶液配制成10.0 mg/mL 混悬液 (以斯皮诺素计),即得。
2.6.2 分组、给药及血样采集[13]18 只大鼠随机分为3 组,给药前禁食不禁水12 h,按20 mg/kg剂量灌胃给予斯皮诺素、固体分散体、磷脂复合物固体分散体混悬液后,于0.5、1、3、5、7、9、10、12、14、16、24 h 眼眶取血各0.3 mL,同时补充适量生理盐水,4 000 r/min 低温离心2 min,血浆置于-20 ℃冰箱中保存。
2.6.3 内标、对照品溶液制备 精密称取盐酸普萘洛尔对照品10 mg 至10 mL 量瓶中,乙腈超声溶解并定容至刻度,流动相稀释至400.0 ng/mL,即得内标溶液。精密称取斯皮诺素对照品10 mg 至50 mL 量瓶中,乙腈超声溶解,室温下放置0.5 h后定容至刻度,得200 μg/mL 贮备液,乙腈稀释至800.0、400.0、200.0、100.0、50.0、10 ng/mL,即得对照品溶液。
2.6.4 样品处理 吸取血浆100 μL、内标溶液(50.0 ng/mL)50 μL 至离心管中,加入1.5 mL 乙腈后涡旋6 min,分层后转移上层有机相,调节氮气吹速后缓慢吹去有机溶剂,100 μL 流动相复溶沉淀物,转移至带内衬管的进样瓶中,待测。
2.6.5 线性关系考察 取“2.6.3” 项下不同质量浓度对照品溶液各500 μL,45 ℃氮气缓慢吹干,加入 空 白 血 浆500 μL、 内 标 溶 液50 μL, 按“2.6.4” 项下方法处理,在“2.1.1” 项色谱条件下进样测定,结果见图4。以对照品溶液质量浓度为横坐标(X),斯皮诺素与盐酸普萘洛尔峰面积之比为纵坐标 (Y) 进行回归, 得方程为Y =0.003 2X-0.151 2 (R2=0.990 9), 在10 ~800.0 ng/mL范围内线性关系良好。

图4 斯皮诺素HPLC 色谱图Fig.4 HPLC chromatograms of spinosin
2.6.6 方法学考察 取10.0 ng/mL (低)、400.0 ng/mL(中)、800.0 ng/mL(高) 质量浓度血浆溶液, 日内精密度 (n =3) RSD 均小于10.66%, 日 间 精 密 度 RSD (n =3) 均 小 于10.26%,表明该方法精密度良好;血浆样品于5 d内在“2.1.1” 项色谱条件下进样测定,测得斯皮诺素、内标峰面积比值无显著差异(P>0.05),表明样品在5 d 内稳定性良好。100 μL 空白血浆配制成10.0、400.0、800.0 ng/mL,在“2.1.1” 项色谱条件下进样测定,将斯皮诺素、内标峰面积比值代入“2.6.5” 项下回归方程,测得3 种质量浓度的方法回收率在86.96%~91.44%之间。
2.6.7 测定结果 在“2.1.1” 项色谱条件下进样测定,通过3P97 程序统计矩模型计算主要药动学参数,结果见表2,再绘制血药浓度-时间曲线,结果见图5。由此可知,固体分散体、磷脂复合物固体分散体主要药动学参数较斯皮诺素均有显著变化(P<0.05,P<0.01);与前者比较,后者tmax显著性延长(P<0.01),Cmax、AUC0~t、AUC0~∞显著升高(P<0.05),两者相对生物利用度分别增加到219.61%、265.39%。
表2 样品主要药动学参数(,n=6)Tab.2 Main pharmacokinetic parameters for samples(,n=6)

表2 样品主要药动学参数(,n=6)Tab.2 Main pharmacokinetic parameters for samples(,n=6)
注:与斯皮诺素比较,△P<0.05,△△P<0.01;与固体分散体比较,*P<0.05,**P<0.01
参数 单位 斯皮诺素 固体分散体 磷脂复合物固体分散体tmax h 5.55±0.88 3.38±0.69△ 5.92±0.93**Cmax ng·mL-1 142.36±36.68 316.51±93.82△△ 406.11±108.02△△*AUC0~t ng·mL-1·h 1 033.75±182.34 2 270.28±426.19△△ 2 743.40±577.27△△*AUC0~∞ ng·mL-1·h 1 046.14±190.68 2 291.65±443.27△△ 2 861.93±597.89△△*
3 讨论
口服制剂进入胃肠道后,需经溶解溶出才能被吸收进入血液循环,从而发挥其药理作用,但难溶性药物溶解度很差,导致其溶出受到极大限制,故提高其溶解度、溶出度成为提高口服吸收生物利用度的关键所在。本实验显示,将斯皮诺素制备成固体分散体后以无定型状态存在,其溶解度、体外溶出度均明显改善, 相对生物利用度增加到219.61%,有助于发挥其药效;与原料药相比,固体分散体tmax显著提前(P<0.05),可能与其促进药物快速溶出有关[7],但磷脂复合物固体分散体tmax与其相比又显著延后(P<0.01),可能是由于它可提高原料药亲脂性[4],导致在胃肠道黏膜中发生了滞留[12-14],同时其相对生物利用度增加到265.39%,Cmax、AUC0~t和AUC0~∞也显著提高。
据报道[14-17],磷脂复合物技术有助于改善药物脂溶性,而将其制成固体分散体后对斯皮诺素水溶性的改善也会产生积极影响,并进一步增加原料药体内吸收。另外,由于磷脂复合物本身黏性较强,故提高斯皮诺素累积溶出度的程度有限,这也印证了继续将其制成固体分散体的必要性。
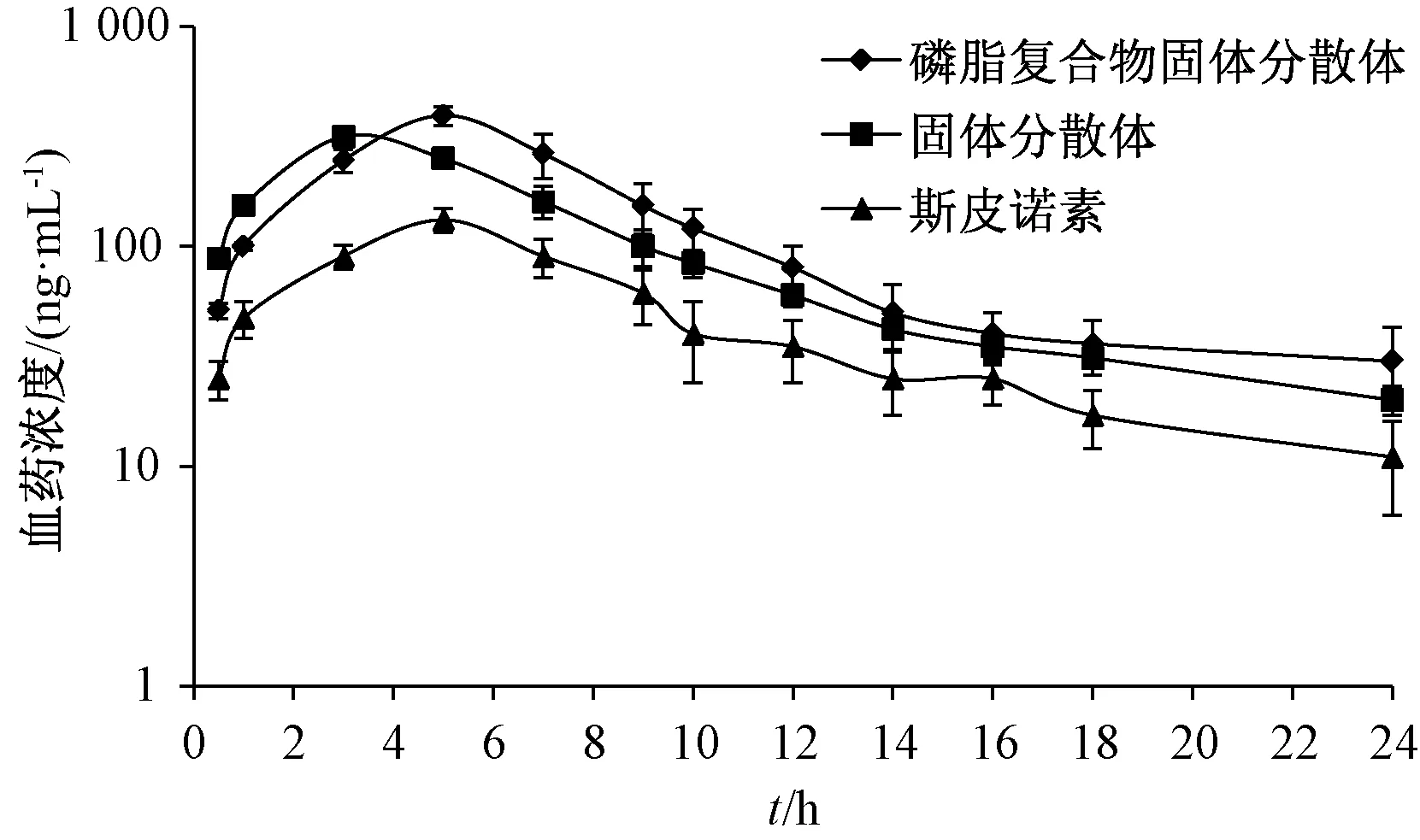
图5 样品血药浓度-时间曲线Fig.5 Plasma concentration-time curves for samples
总之,将磷脂复合物、固体分散体技术联用时,对提高水溶性、脂溶性均较差药物的口服吸收生物利用度具有较好的借鉴意义,本实验也为斯皮诺素今后相关研究奠定基础[18-19]。
猜你喜欢
今日畜牧兽医(2022年10期)2022-12-23
乳业科学与技术(2022年2期)2022-04-15
中国药学药品知识仓库(2022年5期)2022-04-11
中国油脂(2022年1期)2022-02-12
世界科学技术-中医药现代化(2021年5期)2021-11-05
农产品加工(2021年8期)2021-05-20
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
大陆桥视野·下(2017年8期)2017-09-19
试题与研究·中考化学(2016年4期)2017-03-28
中国中药杂志(2016年22期)2017-02-13
