
基于16S rDNA测序对茶园土壤细菌群落多样性的研究
2019-12-31 08:02杨广容马会杰吕才有李永梅
生态学报 2019年22期
杨广容,马 燕,蒋 宾,马会杰,谢 瑾,吕才有,李永梅
1 云南农业大学龙润普洱茶学院,昆明 650201 2 云南农业大学资源与环境学院,昆明 650201
茶树[Camelliasinensis(L.)O.Kuntze]是我国南方重要多年生木本经济作物,2016年,中国茶叶种植面积达293.3×104hm2,约占全球植茶面积的60%以上[1]。茶园土壤环境质量是影响茶叶产量和品质的重要因素之一[2]。茶树在长期连作栽培中不断适应热带和亚热带气候及土壤环境条件,逐渐进化形成了其特有的土壤生态环境特征,如喜温怕冷、喜酸怕碱、喜湿怕涝、喜铵厌硝、聚铝嫌钙、忌氯富锰等。茶树作为典型的喜酸作物,能在pH3.2—6.8的土壤中生长[3-4]。茶园土壤由于茶树长期单一化种植,作物根系分泌物多样性低,施肥、修剪和茶树凋落物归还等原因,致使茶园土壤随茶树种植时间延长而酸化[5-6]。
土壤微生物是土壤中最活跃且多样性最为丰富的组分之一,其群落的组成结构、活性及多样性很大程度上决定了土壤中的营养元素循环和土壤的肥力,是反映土壤质量及评价土壤生态系统可持续性的重要生物学指标,对土壤养分、结构、稳定性上具有重要的影响[5-8]。同时,植被类型和多样性、土壤水分、pH、土壤类型及其理化性状、土地利用方式、有机质、有效磷和速效钾均显著影响微生物群落结构[6,9-11]。如Lauber等认为pH值是决定土壤微生物群落结构的主要因素[12],有研究表明在pH小于6.5的土壤中,土壤微生物多样性随着pH的降低随之降低[13]。已有学者证实,茶园土壤微生物及细菌群落结构受多种生态因子的影响,如:茶树品种及树龄、海拔、季节、施肥及修剪等[5,14-17]。
王秀青等[18]对茶园土壤微生物细菌、真菌和放线菌纯培养研究表明,茶园土壤微生物数量普遍高于森林土壤,并且现代茶园微生物的总数量高于古茶园。细菌是土壤中数量最多、分布最广的微生物,它占土壤微生物总数的70%—90%,其干重约占土壤有机质的1%[4]。由于绝大多数土壤微生物难以培养分离,导致传统研究方法获得的土壤微生物信息较少,不能真实反映土壤微生物的群落结构及功能。采用现代分子生物学研究手段,从土壤中提取微生物宏基因组总DNA,并通过PCR扩增及测序,准确鉴别土壤细菌群落结构及多样性[3-4,19-21],能有效克服传统平板培养计数法的缺陷,也是目前土壤微生物研究的重要内容。本研究以3个古茶园土壤为研究对象,分别以森林和现代茶园土壤为对照,通过直接提取土壤微生物基因组总DNA,采用16S rDNA基因的通用引物PCR扩增及Illumina平台Miseq高通量测序技术,鉴定和比较森林、现代茶园和古茶园土壤细菌的群落结构多样性与差异,为深入了解茶园土壤细菌种群结构演变规律、调节茶园土壤环境质量和改善土壤管理提供理论依据。
1 材料与方法
1.1 土壤采样与处理
1.1.1采样茶园位点概况
选择云南西双版纳州勐海县代表性古茶山景迈山(大平掌)、布朗山(贺开村)和南糯山(半坡竹林小组)的现代茶园(J2、B2和N2)、古茶园(J3、B3和N3)及其附近森林(J1、B1和N1)。各茶园均为生态茶园,除景迈山和布朗山的现代茶园每年进行土壤翻耕外,其他茶园均处于免耕状况,所有茶园栽培管理均为不施肥、不施农药的有机茶园或生态茶园。采样茶园的生态环境概况见表1。
1.1.2土样采集与处理
土样采集时间为2015年5月中旬,云南春茶采摘末期。根据采样茶园的地形条件,在20×20 m区域内,采用3—5点混合取样法,每个采样点沿着茶树树冠边缘垂直投影取样,适当清除地表的枯枝落叶,用土钻采集0—20 cm表层土壤,混合均匀,剔除杂质,形成一个土样,每一茶园重复取3个土样。一部分土样(约20 g)装入无菌袋及时用液氮冷冻后-86℃保存,供分子生物学研究,另一部分土样(约500 g)24 h内室内自然风干,过2 mm或0.15 mm筛用于土壤pH值、阳离子交换量(CEC)、有机质(SOM)、全氮(TN)、全磷(TP)、全钾(TK)、有效氮(碱解氮)、速效磷(Olsen-P)、速效钾及土壤矿质元素有效性钙镁铁铜锌锰的测定。
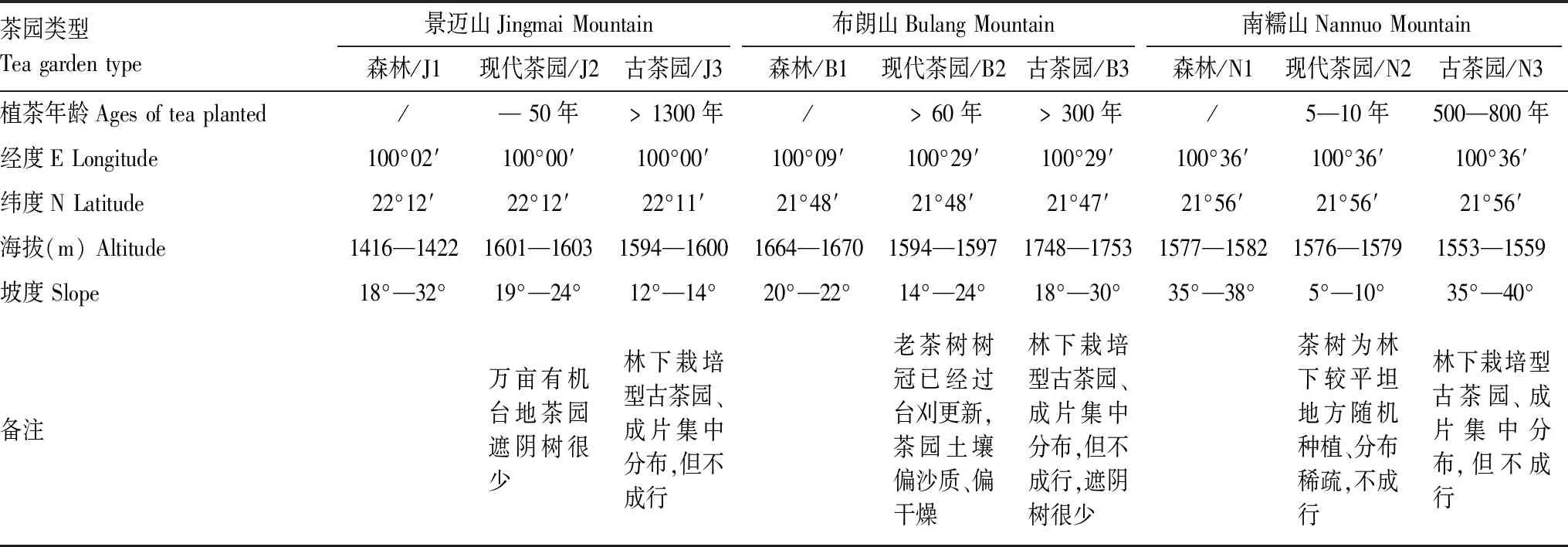
表1 茶园采样位点基本情况Table 1 The basic situation of soil sampling points of tea gardens
1.2 土壤特性及养分含量测定
1.2.1土壤pH值:采用1∶2.5土水(质量)比,用玻璃复合电极法测定。
1.2.2阳离子交换量(cation exchange capacity,CEC)用乙酸铵交换提取—蒸馏法。
1.2.3土壤有机质(soil organic matter,SOM)用重铬酸钾容量法—外加热法。
1.2.4全氮(total nitrogen,TN)用浓硫酸-双氧水消煮—凯氏定氮法;全磷(total phosphorous,TP)用硫酸-双氧水消煮—钼锑抗比色法;全钾(total potassium,TK)浓硫酸-双氧水消煮—火焰光度法。
1.2.5有效氮磷钾:碱解氮(alkali-hydrolyzable nitrogen)用1 mol/L NaOH碱解扩散法;速效磷即Olsen-P用0.5 mol/L NaHCO3溶液浸提—钼蓝比色法;速效钾(available potassium)用乙酸铵浸提—火焰光度法。
1.2.6土壤有效性铜(Cu)、锌(Zn)、锰(Mn)用火焰原子吸收分光光度计法[22]。
各土壤样品的理化性状见表2。
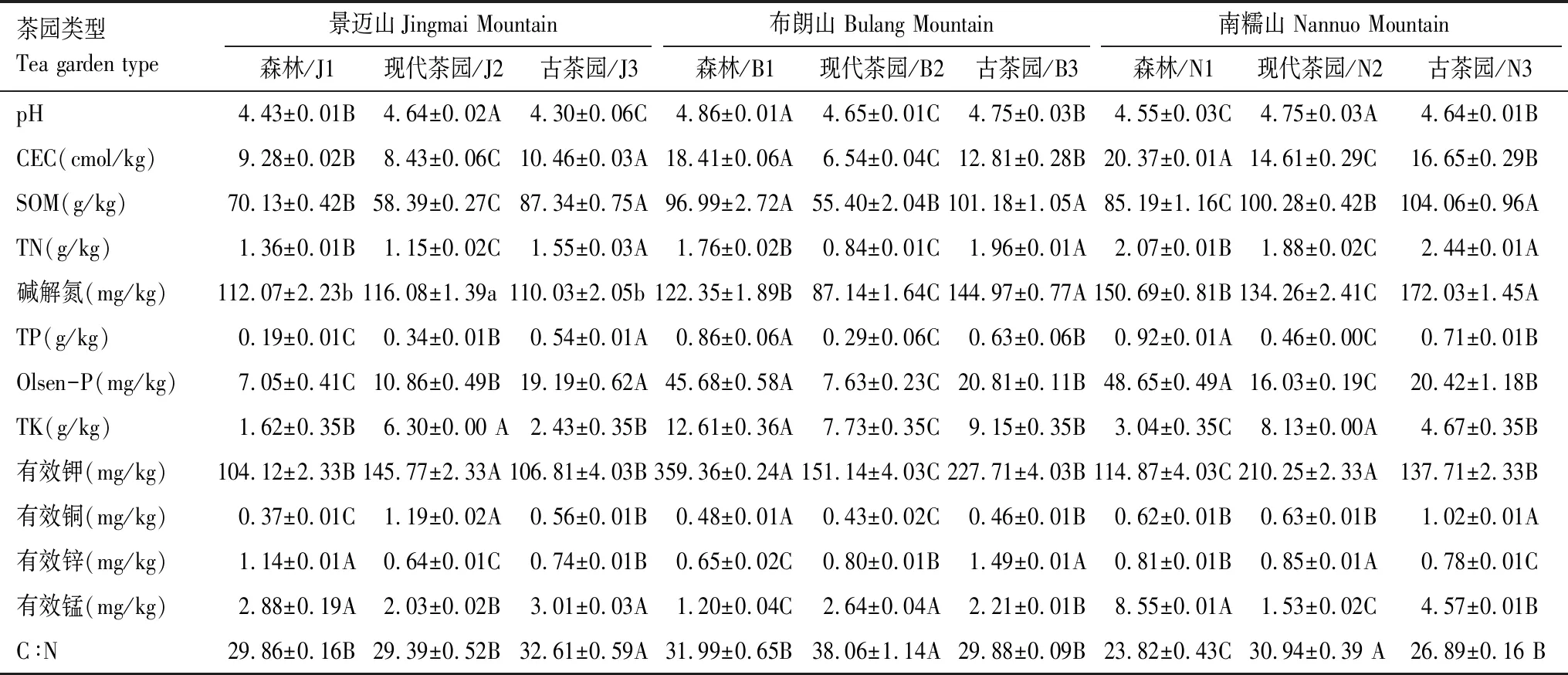
表2 古茶园与现代茶园土壤基本化学性质比较(平均值±标准偏差,n=3)Table 2 The soil physical and chemical properties (means ± standard deviation,n=3)
不同大、小写字母表示同一茶山不同土壤间的差异极显著(P<0.01)和差异显著(P<0.05)
1.3 土壤细菌测序
1.3.1土壤DNA提取
土壤DNA提取参照Proteous等[23]和杨建等[24]的方法,采用SDS-GITC-PEG法,并作适当修改。称取0.5 g土样于2 mL离心管中,所加试剂均按照比例减少20倍,加入氯仿-异戊醇后离心速度改为15 000 g,10 min。其他操作步骤基本不变。
1.3.216S rDNA基因扩增与测序
16S rDNA基因扩增:用16S rDNA基因通用引物341F、806R(见表3)以50 μL体系进行PCR扩增。PCR条件:用带有barcode的特异引物扩增样本16S rDNA特定的V3—V4区域,扩增体系为:50 μL反应体系中包含5 μL的10× KOD Buffer,5 μL的2.5 mmol/L dNTPs,1.5 μL引物(5 μmol/L),1 μL的KOD聚合酶,和100 ng模版DNA。扩增条件为:95℃预变性2 min,随后98℃变性10 s,62℃退火30 s,68℃延伸30 s,共27个循环,最后68℃延伸10 min。

表3 研究中使用的引物Table 3 PCR primers used in this study
测序:对扩增产物切胶回收,用QuantiFluorTM荧光计进行定量。选择符合测序要求的样品16S rDNA送基迪奥生物科技有限公司(广州)对V3和V4区域进行细菌宏基因组测序,将纯化的扩增产物进行等量混合,连接测序接头,根据Illumina官方说明构建测序文库,Hiseq2500的PE250模式上机测序[25]。
数据质量控制:去除测序过程中产生的接头污染序列和一些低复杂度序列(如poly A)及含有N的序列,若reads与接头序列可以连续比对上15 bp的长度,则认为该reads有接头污染;若reads中poly序列的长度≥10 bp,则为低复杂度序列的reads。在进行后续分析之前,还需要对下机数据中质量值较低的碱基质量值小于20的碱基序列进行过滤,保证将一条reads上质量值高于20的碱基数占碱基总数的百分比小于80%的reads过滤掉。
通过重叠关系将双末端测序得到的成对序列组装成一条序列。拼接的条件是最小匹配长度为 50 bp,重叠区域允许的错配率为0.02,符合条件的序列即为目标序列。每个样品测序深度,通过绘制shannon稀释曲线来评价测序量是否足够,并间接反映样品中物种的丰富程度。当曲线趋于平缓或者达到平台期时也就可以认为测序量趋于饱和,基本覆盖样品中所有物种,测序深度增加已经不影响物种多样性。
1.4 数据分析与处理
1.4.1土壤基本性质的分析处理
用SPSS 21.0软件对实验数据进行分析,采用单因素方差分析(oneway-ANOVA)和多重比较(LSD)法进行差异显著性检验和多重比较分析(α=0.05和0.01),图表中的数据均为mean ± SD。
1.4.216S rDNA基因组多样性的标准信息分析
16S rDNA细菌宏基因组V3和V4区域测序结果通过PE reads过滤,获得符合条件的序列即为目标序列tag,利用Mothur[25](v.1.34.0)软件包对tag序列进行了去冗余处理,从中挑选出unique tag序列,并对unique tag的丰度分布进行了统计;使用基于Naive Bayesian方法的分类器rdp classifier工具对tag进行物种注释[26]。根据序列长度的变化,选择恰当的Confidence Threshold(为0.5)参数进行物种分类与丰度分析。
1.4.3细菌宏基因组多样性的高级信息分析
为了更好地获得样品中物种的多样性信息,利用Mothur计算0.03距离下(97%的相似度)的 OTU(Operating Taxonomic Unit)数量及其高级信息OTU丰度、Alpha多样性及聚类等[27-29]。
结合OTU的物种注释信息以及OTU在不同样品中的表达,统计界、门、纲、目、科、属和种每一个分类水平上各样品的表达情况,获得物种分类表达谱,根据物种的分类表达谱的数据,选取丰度较高的部分物种分类单元,用Canoco5.0进行主成分分析(principal component analysis,PCA)。
2 结果与分析
2.1 不同土壤细菌群落丰度及多样性分析
根据细菌16S rDNA的V3—V4区域测序结果,统计9个土壤样品细菌群落丰度及多样性的各项指标(表4)。结果分析表明,所建立细菌文库的覆盖率,除景迈山古茶园土壤(coverage=52.73%)外,其余样本达73.66%—99.55%,平均为83.56%。说明研究所建立的文库可比较真实有效地反映样本环境细菌多样性。森林、现代茶园与古茶园土壤的细菌OTU数目分别为327—17009、5547—25866和10816—27837,表明3座茶山森林土壤的细菌OTU数目差异高达数十倍至数百倍。本研究9个土壤样本细菌群落的Alpha多样性统计表明,chao1和ACE指数变化趋势与OTU指数相一致;景迈山森林(J1)和南糯山森林(N1)与现代茶园(N3)Shannon指数较低,分别为3.17和4.77与4.69,而其他6个土壤的平均Shannon指数达6.98—8.82,说明这6个土壤细菌群落多样性水平较高。一般高肥力土壤中细菌含量较多,本研究3种古茶园和2个树龄较长(50年以上,表1)土壤中细菌含量较为丰富(表4),总体上,同一座茶山古茶园土壤细菌的OTU数、Shannon指数、ACE指数、Chao1指数均高于森林和现代茶园土壤,说明古茶园土壤细菌多样性相对更为丰富。
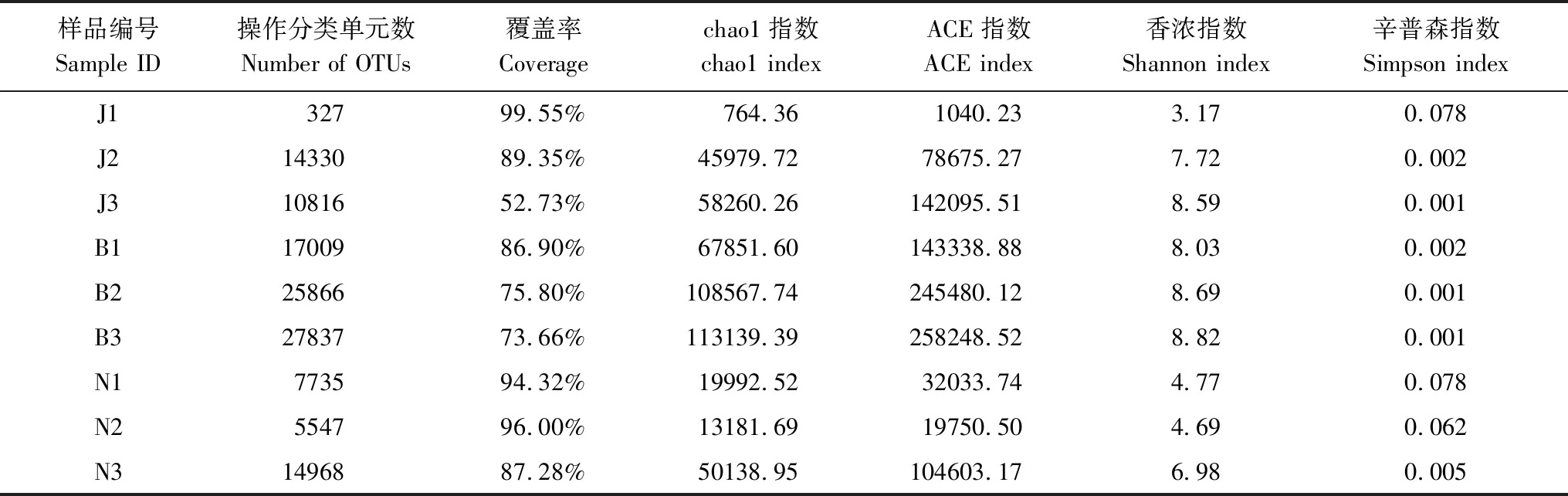
表4 样本细菌Alpha多样性统计Table 4 Statistical analysis of bacterial Alpha deversity in samples
2.2 不同土壤细菌在门水平上的分布
在门分类水平上,经微生物16S rDNA基因测序,检测到样品中共有47个菌门,平均丰度达0.11%及以上的菌门共计21个(表5)。从中可知,不同茶山的森林土壤(J1、B1和N1)及同一茶山的森林、现代茶园和古茶园土壤细菌群落分布差异明显。首先,变形菌门(Proteobacteria)是明显的优势细菌类群,其在9个样品中所占丰度达42.16%—71.85%,平均为50.63%,其次为酸杆菌门(Acidobacteria)、厚壁菌门(Firmicutes)、放线菌门(Actinobacteria)、拟杆菌门(Bacteroidetes)、WPS-2、绿弯菌门(Chloroflexi)、蓝藻细菌门(Cyanobacteria)、浮霉菌门(Planctomycetes)、AD3、疣微菌门(Verrucomicrobia)10个菌门其相对丰度平均达1.06%以上,而其他菌门如芽单胞菌门(Gemmatimonadetes)、泉古菌门(Crenarchaeota)、绿菌门(Chlorobi)、硝化螺旋菌门(Nitrospirae)和梭菌门(Fusobacteria)等10个门的相对丰度低于1.00%。其次,21个主要菌门中,布朗山和南糯山森林土壤(B1和N1)细菌覆盖度达20个菌门,明显高于景迈山森林土壤(J1,仅10个菌门)。最后,除景迈山外,古茶园土壤细菌群落的门类丰度及均匀度均有增加趋势。
表5还表明,9个土壤样本中茶园土壤细菌群落种群分布相对较为稳定,3座茶山现代茶园和古茶园土壤优势菌门变形菌门的平均相对丰度为51.30%和40.92%,其次为酸杆菌门19.61%和19.78%、放线菌门5.46%和7.19%、厚壁菌门4.42%和3.16%与拟杆菌门1.26%和6.03%,此5个菌门相对丰度之和分别为82.48%和77.08%,低于森林土壤5个菌门相对丰度之和的91.86%。表明,植茶和植茶年龄的增加将促进土壤细菌群落优势种群的形成,构成茶园土壤特殊的微生物环境。
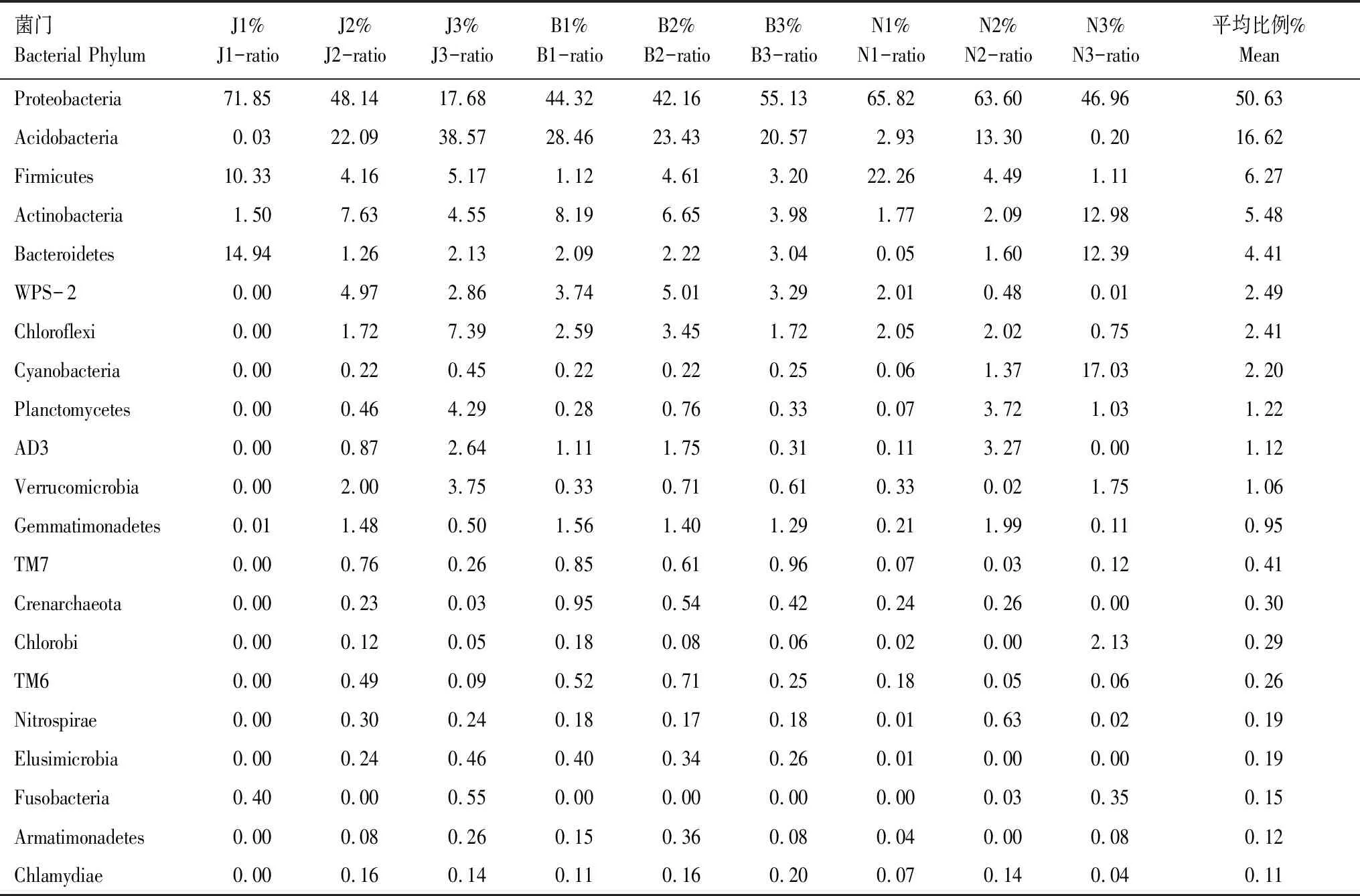
表5 不同土壤细菌在门水平上的分布统计Table 5 Soil bacterial communities at the phylum level
2.3 不同土壤细菌在目水平上的最佳物种分类及聚类分析
为了确定最佳的物种分类水平,统计了9个土壤样品在各个分类水平上的tag序列数及物种分类单元(从上往下)为:界(100%)>门(92.87%)>纲(83.70%)>目(76.76%)>科(44.58%)>属(10.11%)>种(2.00%)。图1为9个样品测序量的OTU稀释曲线及样品间bray距离层级聚类,间接反映样品中物种的丰富程度以及在OTU水平上各样品的相似性。从图1可知,景迈山森林(J1)和南糯山森林(N1)及现代茶园(N2)土壤的OTUs指数(即物种丰富程度)明显低于其他土壤,同时景迈山森林(J1)及古茶园(J3)土壤的OTUs指数(即细菌数量)明显低于其他茶山土壤。布朗山3种土壤(B1、B2和B3)与景迈山现代茶园(J2)土壤细菌群落的物种结构比较相似,而景迈山森林(J1)和南糯山古茶园(N3)土壤的细菌种群结构与其他土壤差异性最大(图1)。
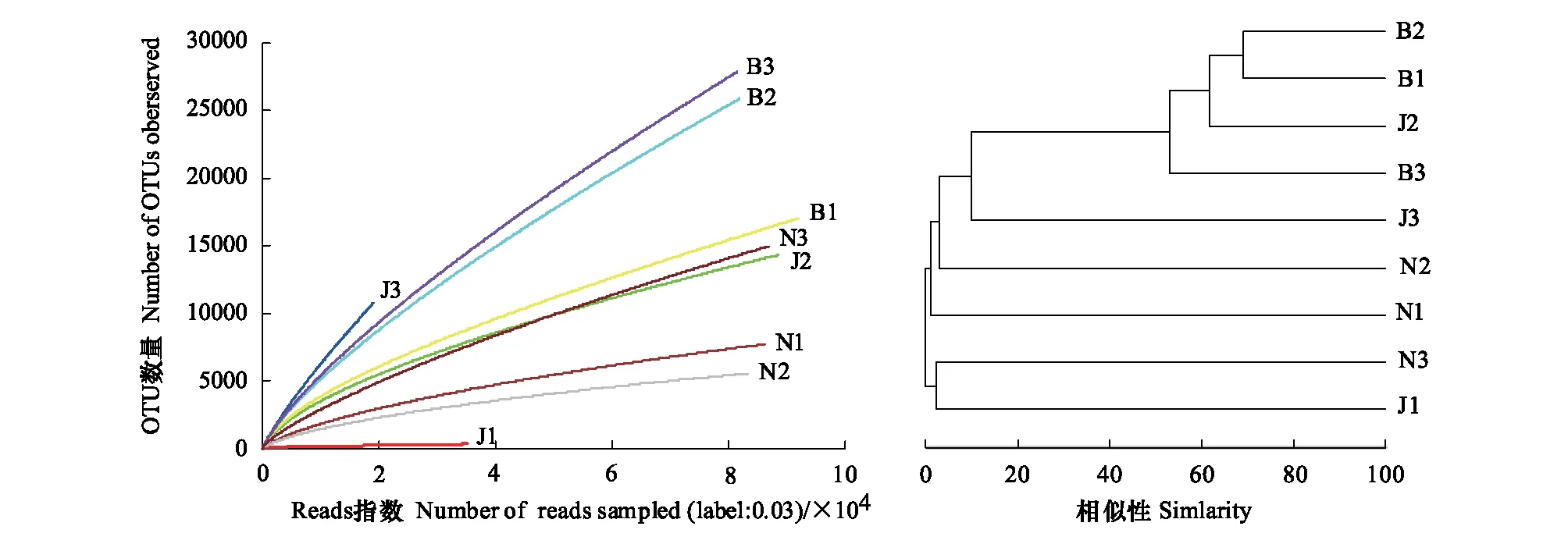
图1 各土壤细菌在97%相似性的OTU稀释曲线图及OTU的bray距离聚类分析图Fig.1 Rarefaction curve at 97% similarity and clustering analysis of OTUs by bray distance
图2是最佳分类水平(目)的物种表达谱热图,它展示了不同的物种在样本间的表达变化情况,热图上的聚类关系,也可以反应样本关系。本研究在目最佳分类水平上总共检测到89个土壤细菌目,它们从属于17个菌门。由于涉及细菌群落的种类很多,图2仅对物种至少在一个样本中包含tags数量达到总tags数量的1%作为阈值的43个目进行作图。结果表明,变形菌门的伯克霍尔德氏菌目(Burkholderiales)和根瘤菌目(Rhizobiales)为森林(J1、B1和N1)、现代茶园(J2、B2和N2)及古茶园(J3、B3和N3)的优势菌目,其在9个土壤样品中的平均丰度分别达13.91%和8.17%,此外,Ellin329、黄单胞菌目(Xanthomonadales)、红螺菌目(Rhodospirillales)、红细菌目(Rhodobacterales)、鞘脂单胞菌目(Sphingomonadales)、柄杆菌目(Caulobacterales)、假单胞菌目(Pseudomonadales)、奈瑟菌目(Neisseriales)和粘球菌目(Myxococcales)的平均丰度分别达6.09%、4.52%、4.40%、3.64%、2.69%、2.20%、1.98%、1.82%和1.56%,变形菌门这11个目的累计平均丰度为50.99%;其次,酸杆菌门的酸杆菌目(Acidobacteriales)、Solibacterales、Ellin6513和iii1-15,其平均丰度分别为9.00%、4.62%、3.64%和1.67%;第三,厚壁菌门的芽孢杆菌目(Bacillales)和乳杆菌目(Lactobacillales)平均丰度分别为4.60%和1.67%,放线菌门的放线菌目(Actinomycetales)和酸微菌目(Acidimicrobiales)则分别为3.46%和1.39%,拟杆菌门的拟杆菌目(Bacteroidales)为2.09%。目最佳分类水平上样品间物种表达聚类结果与图1 OTUs最佳分类水平上的聚类分析结果相一致。
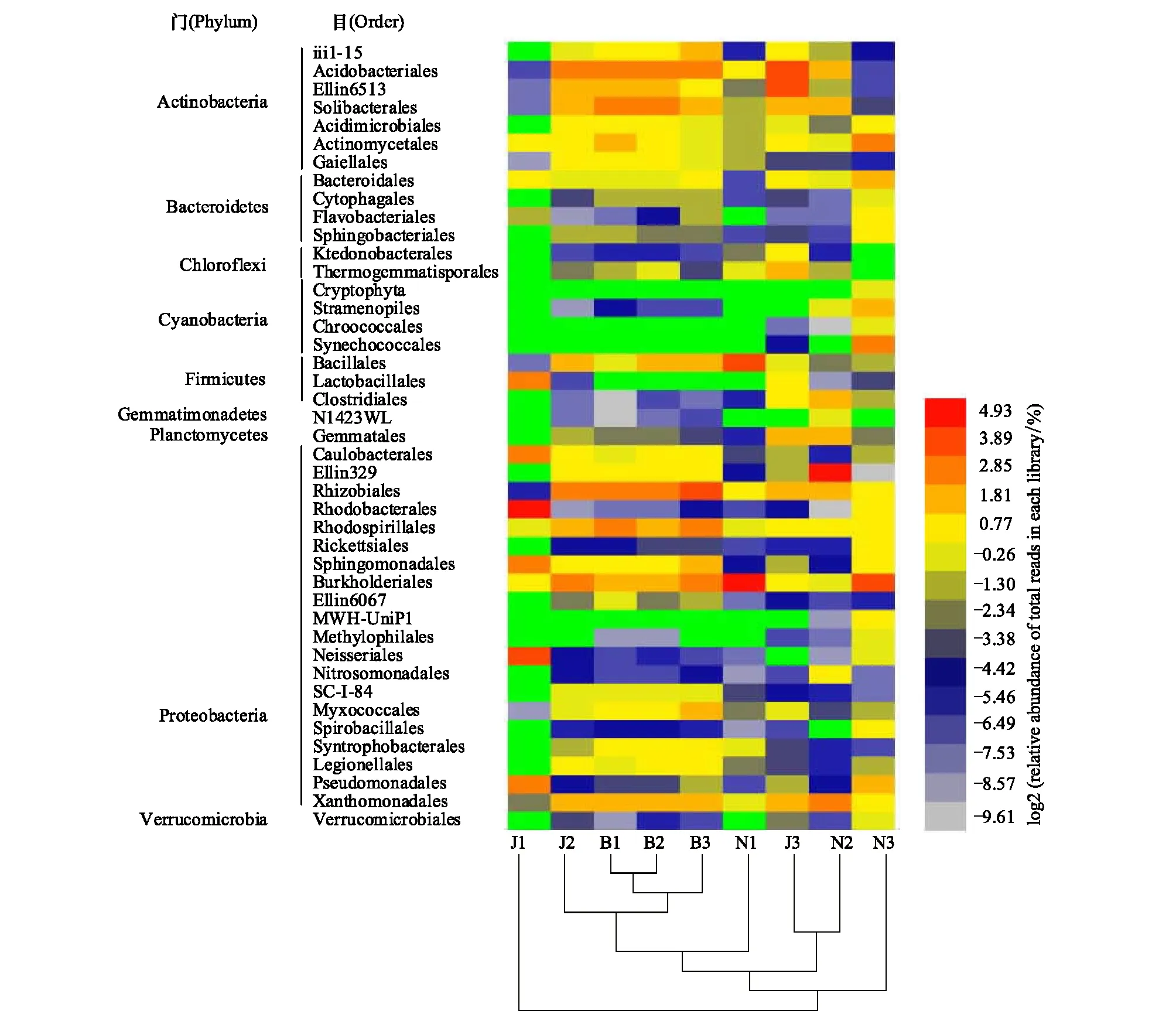
图2 目分类水平各土壤细菌群落结构Fig.2 Soil bacterial communities at order level

图3 各土壤细菌OTU相对丰度的PCA分析Fig.3 PCA analysis of the soil bacterial communities based on the relative abundance of OTU
此外,本研究以OTUs为指标对9个土壤样本进行PCA统计分析(图3),表明:9个土壤PC1=47.62%,PC2=15.97%,布朗山森林(B1)、现代茶园(B2)、古茶园(B3)和景迈山现代茶园土壤(J2)与其他5种土壤细菌种群结构差异可以区分开,南糯山古茶园土壤(N3)也明显区别于其他4种土壤,同理,同一座山不同类型茶园间土壤细菌种群结构也存在明显差异。与此同时,OTUs的PCA分析土壤样本间的相互关系,与上述OTUs最佳分类水平聚类分析(图1)和热图上的物种表达谱的聚类分析(图2)结果相一致。
3 讨论
茶树是广泛适应于热带亚热带酸性土壤的作物,邓欣等[30]和王秀青等[18]采用稀释平板涂抹法等对茶园土壤微生物的数量研究表明,茶园土壤的微生物数量普遍高于森林、农田和旱地土壤。薛冬等[3]采用变性梯度凝胶电泳(DGGE)研究龙井茶产地梅家坞不同茶园、荒地和林地利用方式的土壤微生物群落基因多样性指数表明:荒地 >茶园 >林地。秦红灵等[6]采用RT-PCR分析对红壤坡地不同土地利用方式对土壤细菌数量及结构的影响也表明,茶园 >自然恢复区(不耕种、不施肥,植被自然演替)>农田,茶园土壤细菌数量丰富,并保持较高的多样性。本研究通过对3座茶山森林、现代茶园和古茶园土壤的细菌宏基因组16S rDNA测序及OTUs分子生物信息分析表明:9种土壤样本的细菌数量及Shannon指数、ACE指数、Chao1指数(表4)均为:古茶园 >现代茶园 >森林,说明茶园土壤的细菌种类普遍增加,古茶园土壤细菌多样性相对现代茶园更为丰富,与前人的研究结果基本一致。这可能与茶树长期栽培下,茶园土壤中茶树凋落物、根系分泌物及茶园施肥等使茶园土壤pH值变化及肥力提高(表2)[3-4,14-16,21]及古茶园林下栽培模式下良好的生态环境[18]更有利与茶园土壤细菌的繁衍与生存。
目前国内针对茶园土壤的微生物群落结构的基因组学研究较少,仅卢开阳等[20]采用平板涂布分离法和16S rRNA基因序列分析鉴定南糯山古茶树和台地茶根际土壤细菌群落结构的多样性表明:茶树根际土壤细菌分属于放线菌门、厚壁菌门和变形菌门,其中古茶树生境分离到20个属,台地茶生境则为17个属,在属一级水平,古茶树根际土壤细菌群落的多样性和丰富度均高于台地茶。本研究利用高通量测序技术对森林和茶园土壤细菌群落组成的分子生物信息分析表明:在门分类水平上,9个土壤样本细菌共分属于47个菌门,其中,变形菌门是森林、现代茶园和古茶园土壤的明显优势类群,平均丰度达50.63%,其次,酸杆菌门、放线菌门、厚壁菌门与拟杆菌门,它们和变形菌门相对丰度之和占森林、现代茶园和古茶园的91.86%、82.48%和77.08%。尽管,研究角度和方法不同,但研究结果之间具有很大的相似性,共同表明:茶树栽培管理中,人为干预度较小的林下古茶园栽培模式土壤微生物多样性和丰富度高于人为干预度较大现代茶园。
本研究中关于茶园土壤细菌群落的物种表达热图(图2)说明:3座茶山土壤43个细菌目,从属于9个菌门,其中变形菌门平均丰度达50.99%;其次为酸杆菌门、厚壁菌门、放线菌门及拟杆菌门。这一研究结果与常安然等[31]关于16S rDNA测序技术对烟草根际土壤细菌分类结构研究结果:烟草根际土壤共有25个菌门,其中变形菌门、拟杆菌门、酸杆菌门、疣微菌门四类菌门占明显优势,变形菌门是最为丰富的土壤细菌类群,在不同土壤类型中丰度占39.04%—53.71%相似。此外,国内关于森林及农田土壤细菌多样性的测序研究[32-33]也表明,土壤中优势细菌门为变形菌门、放线菌门、酸杆菌门、厚壁菌门和芽单胞菌门。与其他作物栽培的土地利用方式相比,茶树作为高酚酸含量及根系分泌酚酸较高的多年生作物[3-4],其生物学特性和耕作制度对土壤细菌优势群落在门水平没有显著的影响。但是,从OTUs物种注释聚类分析(图1)和PCA统计结果(图3)则表明,每座茶山森林、现代茶园和古茶园3种土壤类型的细菌群落结构是有明显的差别的。Meta-群落假说认为[34],细菌群落分布主要受内部环境和外部环境两类作用因子的影响,且本土环境特征是调节细菌组成与分布的主要因素。为揭示茶园土壤环境因子和细菌群落组分的内在联系,在今后研究中需要在明确季节、茶园土壤水分、pH值、有机质及速效氮磷钾等因素对细菌群落的影响[6,14-15,31]外,还要进一步扩大茶园土壤微生物研究区域范围、提高土壤微生物基因组的测序深度及物种注释与分类水平,在属或种水平上探明茶园土壤细菌种群结构,才能阐明茶园土壤细菌对土壤环境质量的调控机制。此外,本研究仅对细菌群落进行分析,对于茶园土壤其他种类的微生物类群,如真菌、古菌等的分布结构,还有待于进一步研究。
4 结论
1)土壤细菌16S rDNA高通量测序表明:森林、现代茶园和古茶园土壤细菌群落的平均覆盖率分别为93.59%、87.05%和71.22%,Shannon指数平均分别为5.32、7.03和8.03,表明3座茶山不同土壤的细菌群落丰度为:古茶园 >现代茶园 >森林。
2)在门分类水平上,变形菌门、酸杆菌门、放线菌门、厚壁菌门与拟杆菌门是森林、现代茶园和古茶园土壤的优势类群,它们的相对丰度占比分别达91.86%、82.48%和77.08%;在目最佳分类水平上,变形菌门的伯克霍尔德氏菌目、根瘤菌目为森林和茶园土壤的优势菌种,其平均丰度分别达13.91%和8.17%。
3)3座茶山3种土壤类型的最佳分类水平(目)和OTUs指数PCA统计分析均表明:森林、现代茶园和古茶园土壤类型的细菌群落结构差异明显,除景迈山外,古茶园土壤细菌群落的多样性和丰度均高于森林和现代茶园。
猜你喜欢
当代水产(2022年8期)2022-09-20
昆明医科大学学报(2022年2期)2022-03-29
食品安全导刊(2021年20期)2021-08-30
心声歌刊(2021年6期)2021-02-16
河南科学(2020年3期)2020-06-02
中国比较医学杂志(2020年4期)2020-05-26
湘潮(上半月)(2019年3期)2019-05-22
生物安全学报(2019年3期)2019-02-15
川北医学院学报(2019年6期)2019-02-10
乡村地理(2018年1期)2018-07-06
