
串联的纳米传感器用于癌细胞中miRNA的超灵敏检测
2020-02-06 03:48陈蜜岳仁叶李智王刚林马楠
分析化学 2020年1期
陈蜜 岳仁叶 李智 王刚林 马楠

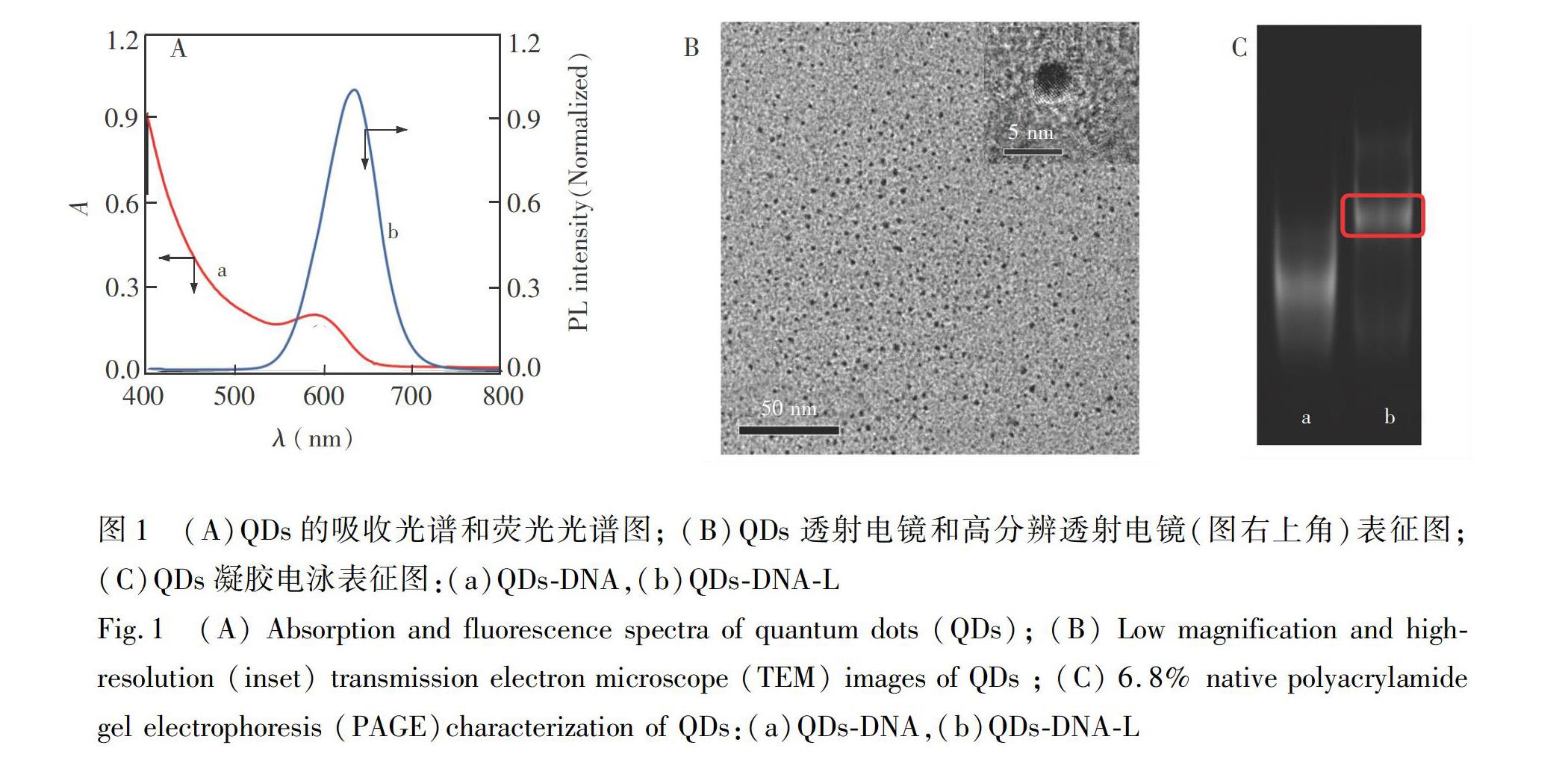
摘 要 MicroRNA(miRNA)的检测在癌症诊断和治疗中具有重要意义。本研究设计了DNA序列,组装了3组金纳米粒子-量子点复合物, 基于此叠加构建了以熵驱动催化循环为模型的单重、双重和多重循环放大的3种纳米传感器。叠加的传感器中,上层复合物催化循环解组装输出的产物作为下层复合物的催化链,引发其解组装,释放更多荧光信号,实现了多重放大。传感器中每叠加一个核酸催化循环体系,检测限降低一个数量级,最终构建的传感器的检测限达到fmol/L级。本研究通过串联熵驱动的核酸催化循环体系,构建了动态可调的、可定量检测细胞中不同表达水平miRNA的新型纳米传感器。
关键词 miRNA; 金纳米粒子; 量子点; 核酸循环; 荧光检测
1 引 言
MicroRNA(miRNA)是一类内源性的小尺寸的非编码RNA分子,长度约为18~25个核苷酸[1~4]。miRNA主要通过与靶基因mRNA配对引导沉默复合体,降解mRNA或阻碍mRNA的翻译[5]。miRNA不仅在生物调节途径中起重要作用[6],也参与肿瘤的发生、发展等病理過程[7]。miRNA的异常表达与癌症的发生密切相关,被认为是诊断癌症的重要生物标志物[8~12]。然而,miRNA序列的高度同源性、尺寸小以及在生物体内含量低等特点[13],使癌细胞中miRNA的精准、灵敏检测极具挑战性。在过去的十几年中,研究者开发了很多技术以提高检测miRNA的灵敏度[14~17],如定量逆转录-聚合酶链式反应(qRT-PCR)技术能灵敏地定量检测低表达的miRNA[17,18],但该方法的仪器设备昂贵且耗时长。
近年来,作为一种有效的信号放大策略,核酸循环为检测低浓度目标物提供了一个重要的技术平台[19,20]。Yin等[21]采用双链特异性核酸酶剪切与miRNA杂交的分子信标放大荧光信号,但核酸酶并不能普遍适用于复杂的生物环境体内; Wu等[22]利用两个发夹DNA间循环催化反应检测细胞内低表达的mRNA,一个靶目标分子经过发夹DNA循环反应后,输出多个荧光信号,但荧光染料光学性能的局限性影响了方法灵敏度。
金纳米粒子(GNP)具有比表面积大、生物相容性好和毒性低等特性,可作为载体将核酸运输至细胞内[23,24],并且对荧光基团有很好的淬灭能力[25]。与传统的荧光染料相比, 量子点(Quantum dots, QDs)具有发射波长可调节、量子产率高、光稳定性好等优点,被广泛应用于生物传感、生物成像领域[26~29]。 Ma等[30]以DNA为模板一步合成生物功能化的水相碲化镉量子点(CdTe QDs),简化了QDs生物功能化的步骤,为构建基于QDs的纳米传感器提供了参考。 Ma等[31]报导了以熵驱动核酸催化循环[32]为模型构建的GNP和QDs组装的纳米传感器,检测细胞内miRNA,检出限达到pmol/L级。尽管对于提高检测miRNA灵敏度的研究取得了一些进展[33,34],但不同癌细胞中miRNA表达水平差异显著,常规传感器的检测限和线性检测区间限制了其在定量分析miRNA中的实际应用。
本研究通过合理设计DNA序列,组装了3组金纳米粒子-碲化镉量子点复合物(GNP-QDs composite, GQC),并依次叠加构建了以熵驱动催化循环为模型的单重、双重和多重循环放大的3种纳米传感器。叠加的传感器中,上层GQC催化循环解组装输出的产物作为下层GQC的催化链,引发其解组装,释放更多荧光信号,实现QDs荧光信号的多重放大。通过叠加多个催化循环体系[35]调控传感器的线性检测区间,降低检测限,用于定量检测不同细胞内不同表达水平的miRNA。
2 实验部分
2.1 仪器与试剂
FE20K酸度计(美国Mettler Toledo公司); AvaSpec-ULS2048-USB2 荧光光谱仪(荷兰Avantes公司); Tecnai G2 F20 S-Twin透射电镜 (美国FEI公司); Agilent 8453 紫外-可见分光光度计(美国 Agilent 公司); GelDoc-It 310 UVP 凝胶成像仪(美国 UVP 公司); Mini-PROTEAN Tetra 垂直电泳装置(美国 Bio-Rad 公司); DYCP-31DN琼脂糖水平电泳仪(北京六一仪器厂); Infinite M200 PRO多功能酶标仪(美国TECAN公司); Nano ZS90 激光粒度仪(英国 Malvern 公司); Olympus IX71 倒置荧光显微镜(日本 Olympus公司)。
氯化镉(CdCl2, 99.99%)、谷胱甘肽(GSH, 98%)、碲粉(Te, 99.99%)、硼氢化钠(NaBH4, 98%)、氯金酸(HAuCl4·3H2O, 99.9%)均购自Sigma-Aldrich公司; 琼脂糖(Agrose,99%)购自Biowest公司; 柠檬酸钠(99%)购自百灵威公司; 三(羟甲基)氨基甲烷(Tris)、过硫酸铵(APS)、MgCl2、NaCl、NaOH、乙酸(HAc)、HNO3、无水甲醇(CH3OH)均至少为分析纯,购自国药集团化学试剂有限公司; HCl购自强盛功能公司; 聚丙烯酰胺(40%)、N,N,N',N'-四甲基乙二胺(TEMED,98%)、甘油(99.0%)购自北京索莱宝公司; 二硫苏糖醇(DTT, 99%)、6-巯基-1-己醇(MCH, 98.0%)购自阿拉丁试剂公司; 三(2-羧乙基)膦盐酸盐(TCEP,98.0%)购自TCI公司; HeLa细胞系购自中国典型培养物保藏中心; 22Rv1细胞系购自上海生物科学研究院细胞资源中心; DMEM培养基、RPMI 1640培养基、1×PBS缓冲溶液、胎牛血清、胰蛋白酶均购自Hyclone公司; miRcute miRNA分离试剂盒购自Tiangen Biotech公司。巯基修饰(Thiolated)DNA购自Takara公司; 荧光染料修饰的DNA购自上海生工生物工程有限公司;硫代DNA以及其它未修饰DNA(序列见表1)购自IDT公司。实验用水为超纯水(Milli-Q Direct8 超纯水系统,美国Millipore公司)。
2.2 实验方法
2.2.1 以硫代DNA为模板一步合成CdTe QDs
按文献\[30\]的方法,用Te和NaBH4制备Te离子的前驱液; 以GSH为配体,与CdCl2配制Cd离子前驱液,与硫代DNA混合后加入Te离子前驱液, 混匀后在100℃下反应,可一步合成DNA4修饰CdTe QDs(Q1)、DNA5修饰CdTe QDs(Q2)和DNA6修饰CdTe QDs(Q3)。用30 kD离心超滤管离心超滤(12500 r/min, 3 min)两次,重新分散于水中。根据文献\[36\]方法计算浓度。
2.2.2 GNP的合成以及表面DNA的修饰 采用柠檬酸还原法合成约13 nm的GNP[32],用 0.22 μm微孔滤膜过滤,紫外可见分光光度计测定520 nm处吸光度, 并计算GNP的浓度(摩尔吸光系数为2.7×108 L/(cm·mol)。 在巯基DNA1、 DNA2和DNA3中加入TCEP, 孵育2 h, 将GNP和巯基DNA按1∶80的摩尔比例混合,37℃恒溫摇荡1 h后离心,加入柠檬酸钠-盐酸缓冲溶液(pH 3),摇荡1 h,12500 r/min离心7 min,分散在1×PBS中,得到GNP -DNA1(G1)、GNP-DNA2(G2)和GNP -DNA3(G3)。
2.2.3 GQC的组装 将G1与L1按摩尔比1∶60混合,加入NaCl(终浓度50 mmol/L)和MgCl2(终浓度2.5 mmol/L),于37℃反应过夜,12000 r/min离心6 min,分散在1×PBS缓冲液中。将组装好的G1-L1与Q1按照GNP与QDs摩尔比为1∶60混合,加入NaCl(终浓度50 mmol/L)和MgCl2(终浓度2.5 mmol/L) 37℃反应3 h,缓慢降至室温。12000 r/min离心7 min,分散于1×PBS缓冲液中,得到复合物GNP-QDs1(GQC1)。按同样步骤,分别通过L2和L3将G2和Q2、G3和Q3组装成GQC2和GQC3。
2.2.4 GNP修饰DNA数量的测定 将序列与DNA1一致的FAM-DNA1按照2.2.2节的步骤修饰在GNP表面,取100 μL FAM-DNA1-GNP(36.5 nmol/L)与80 μL DTT(10 mmol/L)混合,振荡12 h,14000 r/min离心5 min,收集上清液,酶标仪测定荧光强度,激发波长480 nm,发射波长520 nm。酶标仪测定浓度分别为10、20、30、40和50 nmol/L 的FAM-DNA1荧光强度,绘制标准曲线,通过标准曲线计算GNP表面连接的FAM-DNA1的浓度,与GNP浓度的比值即为GNP表面修饰DNA1的数量。GNP表面修饰的DNA2和DNA3的数量按照以上方法测定。
将与L1序列一致的FAM-L1与G1按照2.2.3节组装,测定GNP连接的L1条数。采用同样方法测定和计算GNP表面连接的L2和L3的数量。
2.2.5 纳米复合物特异性测试
用1×PBS缓冲液配制一系列含5 nmol/L GNP 的GQC1溶液,并与 F1(终浓度60 nmol/L)混合,加入miR-21(终浓度为100、10、2、1、0.2、0.1和 0 nmol/L); 对照组(C' control)的GQC1中加入终浓度为100 nmol/L miR-141和 60 nmol/L F1, 37℃反应6 h,12000 r/min离心3 min,取上清液测荧光信号。
2.2.6 纳米传感器的构建和循环检测DNA 由GNP浓度为5 nmol/L GQC1构建传感器C1; 由GNP浓度均为5 nmol/L GQC1和GQC2混合(即串联),构建传感器C2; 由GNP浓度分别为5、5和10 nmol/L的 GQC1、GQC2和GQC3混合,构建传感器C3。
在3组传感器C1、C2和C3中加入对应的fuel DNA(终浓度分别是60 nmol/L F1; 60 nmol/L F1和60 nmol/L F2; 60 nmol/L F1、60 nmol/L F2和120 nmol/L F3),加入靶分子C'至终浓度分别是100、10、2、1、0.5、0.2、0.1、0.05、0.02、0.01、0.005、0.002、0.001和0 nmol/L,在37℃分别反应6、8和12 h。离心,测定上清液荧光信号,下层沉淀用1×PBS缓冲液分散后,进行琼脂糖凝胶电泳分析。
2.2.7 固定细胞成像 在48孔板中接种HeLa细胞和22Rv1细胞,每孔细胞数约2×104个,在细胞培养箱中37℃孵育24 h。弃去培养液,细胞用甲醇固定,加入传感器C1、C2和C3以及相应的Fuel DNA,培养箱中37℃孵育6、8和12 h,用1×PBS缓冲液清洗细胞,采用荧光倒置显微镜成像。
2.2.8 提取RNA定量检测miRNA 在细胞培养瓶中分别接种HeLa细胞和22Rv1细胞,待2种细胞长满培养瓶后,采用试剂盒提取细胞的总RNA,测量总RNA在260 nm处的吸光度(1 OD=40 ng/μL RNA)。将提取的RNA稀释,与相应FuelDNA混合后加入对应的传感器中,测定荧光光谱,计算荧光恢复率,并根据线性曲线计算miR-141在两种细胞中的表达量。
3 结果与讨论
3.1 QDs和GNP组装和表征
DNA功能化CdTe QDs的吸收光谱和荧光谱如图1A所示, QDs的最大吸收峰位于571 nm(曲线a),最大发射波长为627 nm(曲线b), 计算QDs的量子产率为17.6%[32]。采用透射电镜和高分辨透射电镜对QDs进行表征(图1B), QDs直径为(3.43±0.18) nm。QDs进行凝胶电泳图谱见图1C,QDs表面大多修饰了一条DNA链。
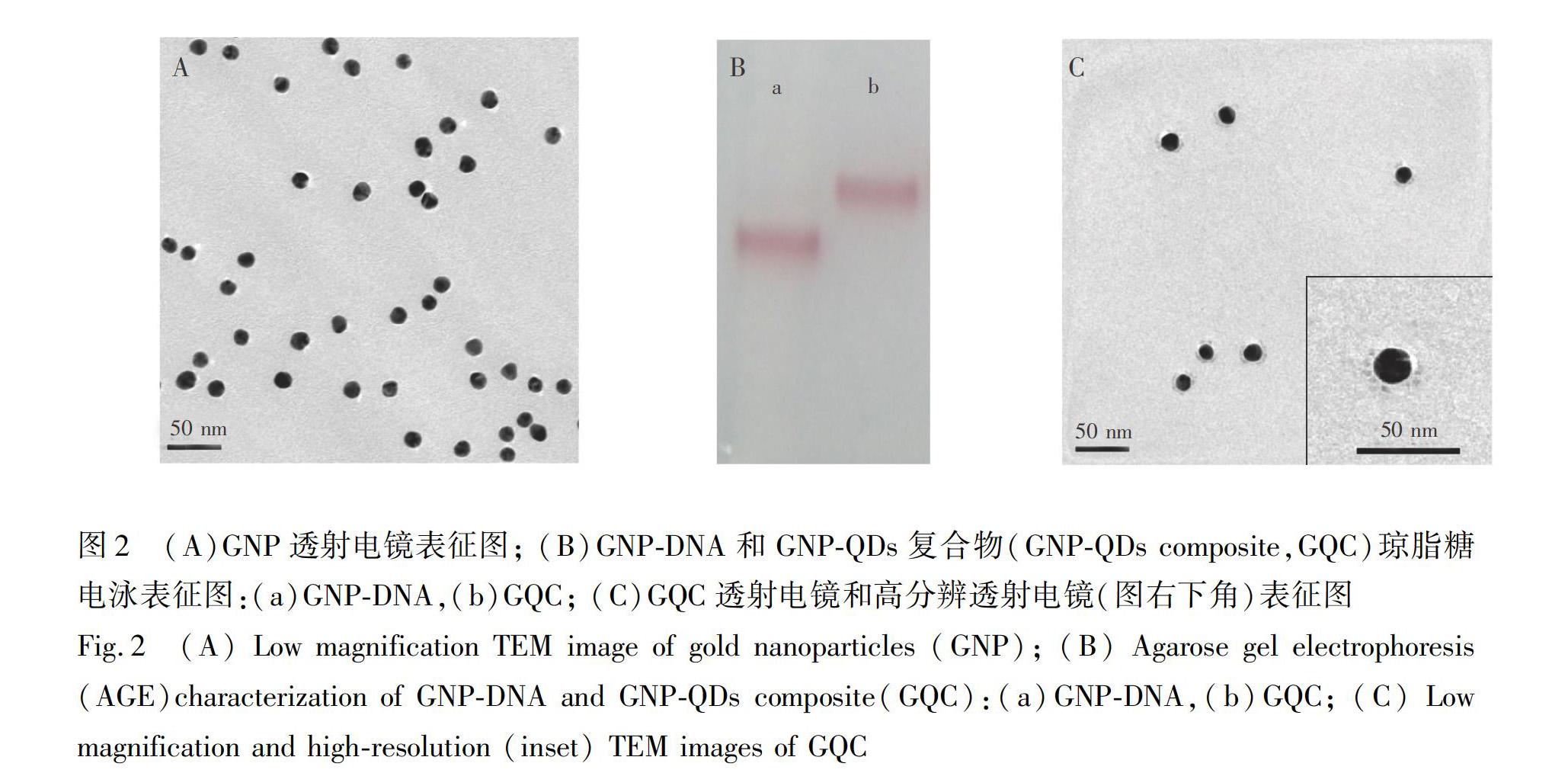
GNP电镜表征图如图2A所示,其直径为(15.48±0.23) nm。使用DTT法测得每个GNP表面修饰了36条巯基DNA和20条Linker DNA。琼脂糖电泳表征图(图2B)和电镜表征图(图2C)都表明GNP和QDs形成了组装体,且每个GNP表面组装了约12个QDs[32]。
3.2 串联传感器的构建以及可行性验证
本研究以熵驱动核酸催化循环为模型构建纳米传感器,实现检测信号的放大。在传感器C1中,Q1通过L1与G1组装成GQC1,由于荧光共振能量转移作用,Q1的荧光被G1猝灭,miR-141作为GQC1的靶分子,进攻作用位点Toehold 1,将G1取代,由于G1与Q1距离增大,不能发生荧光共振能量转移,Q1的荧光信号恢复。同时,作用位点Toehold 2暴露,F1与之结合,将Q1从组装中取代出来,同时又置换出miR-141,miR-141继续进攻下一个作用位点Toehold 1,释放Q1,如此循环下去,一个靶目标循环解组装GQC1输出多个Q1荧光信号(图3A)。将GQC1和GQC2按1∶1比例叠加构建传感器C2,miR-141作为靶向物催化解组装GQC1中置换出的Q1作为GQC2的催化链,与F2通过Toehold 3和Toehold 4两作用位点,按照上述循环方式继续催化解组装GQC2,释放Q2。如此,在传感器C2中输入一个靶目标分子,能够输出多个Q1和Q2的荧光信号; 将GQC1、GQC2和GQC3按1∶1∶2比例叠加构建传感器C3,GQC2中被Q1催化解组装输出的Q2又作为GQC3的催化链,与F3通过Toehold 5和Toehold 6两作用位点催化解组装GQC3,释放Q3,如此,在传感器C3中,输入一个靶目标能够输出多个Q1、Q2和Q3的荧光信号(图3B),检测靶目标miR-141对应的荧光信号,得到多重放大。
采用凝胶电泳表征单链DNA间杂交、链取代反应,以验证该实验设计的可行性。如图4所示,泳道g对应在GQC2的单链组装体加入对应的催化链(DNA4)和F2后解组装的条带; 泳道h对应将GQC1和GQC2的单链组装体1∶1串联,只加入了GQC1的靶目标(0.05×C')和F1和F2的情况; 泳道h中同样也出现了GQC2单链组装体解组装的条带, 并且条带的荧光强度比泳道g中的条带的荧光强度高,表明将核酸循环串联可实现信号的多重放大。
考察了GQC1对miR-141的特异性响应。将miR-21按照与L1(100 nmol/L)摩尔比分别为1、0.1、0.02、0.01、0.002、0.001和0的比例,与F1加入至C1传感器中,与对照组(C' control)相比,GQC1的荧光强度基本没有变化(图5),即没有QDs被miR-21从GQC1解组装出来,表明此复合物可特异性识别miR-141。
3.3 串联传感器循环检测miRNA
探究3组传感器C1、C2和C3对不同浓度的靶向物C'(miR-141)的响应能力,将与L1(100 nmol/L)不同摩尔比(1、01、002、001、0005、0002、0001、00005、00002、00001、000005、000002、000001和0)的C'和對应的Fuel加入至C1、C2和C3传感器中,采用琼脂糖电泳和荧光光谱表征复合物解组装的程度。
如图6所示,随着C'/L1的比例不断增加, 复合物解组装越完全,在泳道中迁移速率越快。传感器C1、C2和C3分别对在0001~1、00001~1和000001~1(C'/L1摩尔比)范围内对靶向物有响应。随着核酸循环体系的叠加,对应的传感器的灵敏度也随之提高,可响应的靶向物浓度更低。因此,可通过改变叠加的循环体系的数量调控靶向物检测范围。
图7A~7C是由不同比例的C'/L1分别滴定C1、C2和C3传感器释放的QDs的荧光谱图,在不同C'/L比例下传感器解组装释放的QDs荧光强度与琼脂糖电泳表征的解组装结果基本一致。图7D为3组传感器中QDs荧光恢复率(F/F0-1)与不同比例C'/L1的关系图, 根据图7D将3组传感器检测的C'/L1摩尔比例区间转化为C'浓度区间,其中C1为10-10~10-7 mol/L,C2为10-11~10-7 mol/L,C3为10-12~10-7mol/L,可见每叠加一个核酸催化循环体系,其对应的传感器最低检测范围降低一个数量级。
根据图8中3组传感器的线性检测曲线,计算C1、C2和C3的检测限分别为47.3 pmol/L、4.23 pmol/L和552 fmol/L(3σ/k)。
3.4 固定细胞成像
miR-141在22Rv1细胞中表达含量高, 在HeLa细胞中表达含量较低[21,37]。将C1、C2和C3传感器和对应的Fuel与上述两种固定细胞分别孵育6、8和12 h后的细胞荧光成像图如图9所示, 22Rv1细胞与3组传感器孵育后均有明显的荧光成像信号, 并随着每一次核酸循环体系的叠加, 荧光信号强度也随之增强; C1传感器在HeLa细胞中并没有明显的QDs荧光成像信号,这是因为HeLa细胞中miR-141表达含量比较低[38], 其含量低于C1传感器检测限。选取相应的传感器定量检测两种细胞中miR -141的含量(表2),并与文献对比发现,提取RNA定量检测miR -141的拷贝量与文献报导的值相接近[21,37~39]。
4 结 论
通过将核酸循环体系叠加,开发了新型纳米传感器,与常规的纳米传感器相比,串联的纳米传感器具有动态可调的线性检测区间和检测限,可用于灵敏检测癌细胞中不同表达水平的miRNA分子。虽然通过串联核酸循环能够放大检测信号,但伴随着信号的放大,传感器的背景信号也随之增大,影响串联传感器的灵敏度的提高。 后续研究将对传感器背景信号增加的原因进行分析,进一步提高串联传感器的灵敏度。
References
1 Wienholds E, Plasterk R H. FEBS Lett., 2005, 579(26): 5911-5922
2 He L, Hannon G J. Nat. Rev. Genet., 2004, 5(7): 522-531
3 Ambros V. Nature, 2004, 431(7006): 350-355
4 Dong H, Lei J, Ding L, Wen Y, Ju H, Zhang X. Chem. Rev., 2013, 113(8): 6207-6233
5 Bartel D P. Cell, 2009, 136(2): 215-233
6 Bartel D P. Cell, 2004, 116(2): 281-297
7 Di Stefano V, Zaccagnini G, Capogrossi M C, Martelli F. Vasc. Pharmacol., 2011, 55(4): 111-118
8 Esquela-Kerscher A, Slack F J. Nat. Rev. Cancer, 2006, 6(4): 259-269
9 Calin G A, Croce C M. Nat. Rev. Cancer, 2006, 6(11): 857-866
10 Liang D, Meyer L, Chang D W, Lin J, Pu X, Ye Y Q, Gu J, Wu X F, Lu K. Cancer Res., 2010, 70(23): 9765-9776
11 Witwer K W. Clin. Chem., 2015, 61(1): 56-63
12 Kayano M, Higaki S, Satoh J I, Matsumoto K, Matsubara E, Takikawa O, Niida S. Biomarker Res., 2016, 4(1): 22
13 Murakami Y, Yasuda T, Saigo K, Urashima T, Toyoda H, Okanoue T, Shimotohno K. Oncogene, 2006, 25(17): 2537-2545
14 Sato Y, Ichihashi T, Nishizawa S, Teramae N. Angew. Chem. Int. Ed., 2012, 51(26): 6369-6372
15 Cheung T H, Quach N L, Charville G W, Liu L, Park L, Edalati A,Yoo B, Hoang P, Rando T A. Nature,2012, 482(7386): 524-528
16 Hansen T B, Jensen T I, Clausen B H, Bramsen J B, Finsen B, Damgaard C K, Kjems J. Nature, 2013, 495(7441): 384-388
17 Benes V, Castoldi M. Methods, 2010, 50(4): 244-249
18 Marabita F, de Candia P, Torri A, Tegner J, Abrignani S, Rossi R L. Brief. Bioinform., 2015, 17(2): 204-212
19 Wang F, Lu C H, Willner I. Chem. Rev., 2014, 114(5): 2881-2941
20 Chen J, Tang L, Chu X, Jiang J. Analyst, 2017, 142(17): 3048-3061
21 Yin B C, Liu Y Q, Ye B C. J. Am. Chem. Soc., 2012, 134(11): 5064-5067
22 Wu C, Cansiz S, Zhang L, Teng I T, Qiu L, Li J, Tan W. J. Am. Chem. Soc., 2015, 137(15): 4900-4903
23 Khlebtsov N, Dykman L. Chem. Soc. Rev., 2011, 40(3): 1647-1671
24 Seferos D S, Giljohann D A, Hill H D, Prigodich A E, Mirkin C A. J. Am. Chem. Soc., 2007, 129(50): 15477-15479
