
孔雀石绿分子印迹电化学传感器的制备与应用
2020-02-06 03:48韦寿莲吴嘉喻黄象金谢春生
分析化学 2020年1期
韦寿莲 吴嘉喻 黄象金 谢春生


摘 要 以孔雀石绿(MG)为模板分子,通过电聚合邻苯二胺,在多孔碳纳米纤维(PCNF)修饰的玻碳电极表面制备了可特异性识别MG的分子印迹传感器。采用差分脉冲伏安法(DPV)、循环伏安法(CV)和电化学阻抗法(EIS)表征了传感器的电化学性能,优化了PCNF的滴涂量、模板分子与功能单体的摩尔比、CV扫描圈数、洗脱剂、洗脱时间、样品溶液pH值及孵化时间等实验参数。在最佳条件下,DPV峰电流强度与MG浓度在
0.10~10.0 nmol/L范围内呈良好的线性关系,检出限为0.042 nmol/L(3S/k)。此传感器具有良好的选择性、重现性和稳定性。将本方法应用于市售新鲜草鱼及养殖水样品中MG的含量测定,加标回收率为82.0%~96.5%。
关键词 分子印迹电化学传感器; 电聚合; 多孔碳纳米纤维; 孔雀石绿
1 引 言
孔雀石绿(Malachite green, MG)是一种阳离子三苯甲烷染料,因其对鱼类的真菌和寄生虫感染有很好的抑制作用,曾在水产养殖中被用作抗菌和抗寄生虫药[1]。MG很容易被鱼组织吸收,并大部分被代谢成亲脂的隐色孔雀绿,它在鱼组织中可持续存在很长时间[2]。MG及其代谢物对人类具有致畸、致癌和致突变作用,许多国家禁止在水产养殖和水产品中使用MG[3,4]。但因MG的抗菌效果显著、价格低,在一些地区仍被用于水产养殖,给水体、鱼类产品和人体健康造成危害。因此,建立灵敏、准确、简便的MG检测方法具有重要的意义。
目前,MG的检测方法包括高效液相色谱[5,6]、液相色谱-质谱联用技术[7,8]、毛细管电泳法[9]、表面增强拉曼光谱法[10]和电化学方法[11,12]等。其中电化学方法具有响应快、灵敏度高、操作简单和易于微型化等优点,特别适合于常规分析。分子印迹聚合物(Molecularly imprinted polymers, MIPs)是一种人工合成聚合物,拥有与模板分子的尺寸、形状和功能基团互补的印迹位点,对模板分子具有高亲和力和选择性,因其具有稳定性好、可重复利用、造价低廉等优点,在电化学传感领域具有良好的应用前景[13~15]。已有文献报道将分子印迹技术与电化学分析法相结合用于不同样品中痕量MG的高选择性测定。如Huang等[16]开发了一种分子印迹电化学发光法,用于检测鱼类样品中的微量MG,检出限为7.3 ng/kg。尽管MIPs在提高选择性方面具有很好的优势, 但在电化学检测中其灵敏度较低。因此,各种纳米材料被用于修饰电极以提高电极的有效比表面积、电子传递速率和电催化活性[17],从而提高分子印迹电化学传感器的检测灵敏度。纳米碳纤维具有稳定性好、比表面积大、电导性高等优良性能,在电化学分析中受到越来越多的关注[18,19]。尤其是以细菌纤维素为碳源制备的多孔碳纳米纤维(Porous carbon nanofiber, PCNF), 因其较强的机械强度、高孔隙率和廉价易得而备受关注,并且已被作为电极修饰材料应用于重金属离子的高灵敏电化学检测[20]。
本研究以细菌纤维素为碳源制备了PCNF材料,并以其作为电极增敏材料。以MG为模板分子,邻苯二胺为功能单体,通过电聚合法在PCNF修饰的玻碳电极上合成MG的MIPs,构建了MG分子印迹电化学传感器,并用于实际样品中MG的测定。
2 实验部分
2.1 仪器与试剂
透射电子显微镜(Transmission electron microscope, TEM, 荷兰Philips公司); Tristar 3000比表面积分析仪(BET, 美国Micromeritics公司); CHI660E电化学工作站(上海辰华仪器公司)。采用三电极体系,玻碳电极(Glassy carbon electrode, GCE, 直径为2 mm)或其修饰电极为工作电极, 铂丝为对电极, 饱和甘汞电极为参比电极。
MG标准品(上海阿拉丁试剂有限公司); 乙腈(色谱纯,上海安谱公司); 邻苯二胺、K3[Fe(CN)6](分析纯,广州化学试剂厂); 酸性Al2O3、盐酸羟胺(分析纯,天津市大茂化学试剂厂); 其它试剂均为分析纯。实验用水为超纯水。0.2 mol/L PBS缓冲溶液由NaH2PO4和Na2HPO4配制; 0.05 mol/L乙酸铵缓冲溶液由乙酸铵和乙酸配制; 0.2%(w/w)壳聚糖溶液由1%(V/V)乙酸溶液配制。
2.2 实验方法
2.2.1 PCNF的制备 使用超纯水将细菌纤维素洗至中性,冷冻干燥24 h。将干燥后的细菌纤维素放至管式炉中,在氮气保护下进行程序控温热处理,升温速率为10℃/min,在800℃下碳化2 h,得到PCNF材料。
2.2.2 分子印迹电极及非印迹电极的制备
在麂皮上用0.5 μm的Al2O3粉末将玻碳电极打磨抛光,然后依次用无水乙醇、水冲洗干净,晾干。称取5 mg PCNF于5.0 mL 0.2%壳聚糖溶液中,超声混匀,制得1.0 mg/mL的PCNF分散液。吸取4.0 μL PCNF分散液滴涂于處理好的玻碳电极表面,在红外灯下烘干,即得到PCNF修饰电极(PCNF/GCE)。将PCNF/GCE置于10 mL PBS缓冲液(pH 7.0)中,此缓冲液含有1.0 mmol/L MG和4.0 mmol/L邻苯二胺。以PCNF/GCE为工作电极,铂丝为对电极,饱和甘汞电极为参比电极,在0~0.8 V电压范围内,以100 mV/s循环扫描20圈,形成MG印迹膜。将聚合后的电极置于80%(V/V)的甲醇-乙酸混合液中搅拌洗脱25 min,去除模板分子,即获得MG分子印迹电化学传感器(MIPs/PCNF/GCE)。非印迹电化学传感器(NIPs/PCNF/GCE)按上述同样步骤制备,但不加MG。
2.2.3 电化学测试 将印迹电极在MG溶液中孵育20 min,取出电极,用超纯水冲洗电极表面,采用电化学测试。每次测试完成后将电极置于20%(V/V)的乙酸-甲醇溶液中进行洗脱25 min,再用水冲洗干净,待继续使用。差分脉冲伏安法(DPV)、循环伏安法(CV)和电化学阻抗(EIS)测试采用三电极体系,在含0.10 mol/L KCl的5.0 mmol/L K3[Fe(CN)6\]溶液中进行。DPV扫描电位为-0.2~0.6 V,电位增量为4 mV,振幅为50 mV,脉冲周期为0.5 s; EIS频率范围为0.1 Hz~100 kHz,振幅为5 mV。
2.2.4 样品制备 养殖水样取自肇庆市周边的鱼塘。水样经0.22 μm滤膜过滤后进行电化学检测。 草鱼从肇庆当地市场购买。鱼去皮,取可食部分切片,匀浆。称取5.00 g样品于50 mL离心管中,依次加入1.5 mL 0.25 g/mL盐酸羟胺溶液、3.5 mL 0.05 mol/L乙酸铵缓冲溶液(pH 4.5),涡旋30 s。然后加入10 mL乙腈涡旋提取10 min,再加入5 g酸性Al2O3和2 g NaCl,涡旋2 min,以5000 r/min离心10 min, 收集上清液。再用10 mL乙腈重复提取残渣,合并上清液,于40℃下氮气吹至近干,用纯净水溶解,定容至10.0 mL,经0.22 μm滤膜过滤后,采用本方法进行检测。
3 结果与讨论
3.1 PCNF的表征
由PCNF的透射电镜图(图1A)可见,PCNF的直径为20~80 nm,大量PCNF相互交错形成多孔的网状结构,这些互通的孔隙为电解质离子提供了充足的扩散通道。N2吸脱附等温曲线见图1B,此N2吸脱附等温线属于典型的IV类曲线,在相对压力为0.45~0.97下具有明显的H2滞后环,说明PCNF存在介孔结构; 在较高的相对压力(0.97)下,曲线尾部急剧上升,这是由于PCNF间形成的大孔中N2的多层吸附引起的[21],这些大孔可在透射电镜图中观察到。根据N2吸脱附等温曲线计算得到PCNF的比表面积为687 m2/g。由图1B插图可知,PCNF具有窄的孔径分布,集中在3 nm附近。
3.2 电聚合制备分子印迹膜
图2为含MG的聚合液在PCNF/GCE表面电聚合过程的CV曲线。由CV曲线可知,邻苯二胺的电聚合是一个不可逆的氧化过程,峰电流随着电聚合圈数增加而显著减小,这是由于电极表面形成了致密的不导电聚合膜,阻碍了电极与溶液之间的电子转移。插图为不加MG的邻苯二胺电聚合的CV曲线,与电聚合制备MIPs的 CV曲线相比无明显差异,说明MG在0~0.8 V电聚合过程中无电活性。
3.3 分子印迹传感器的电化学表征
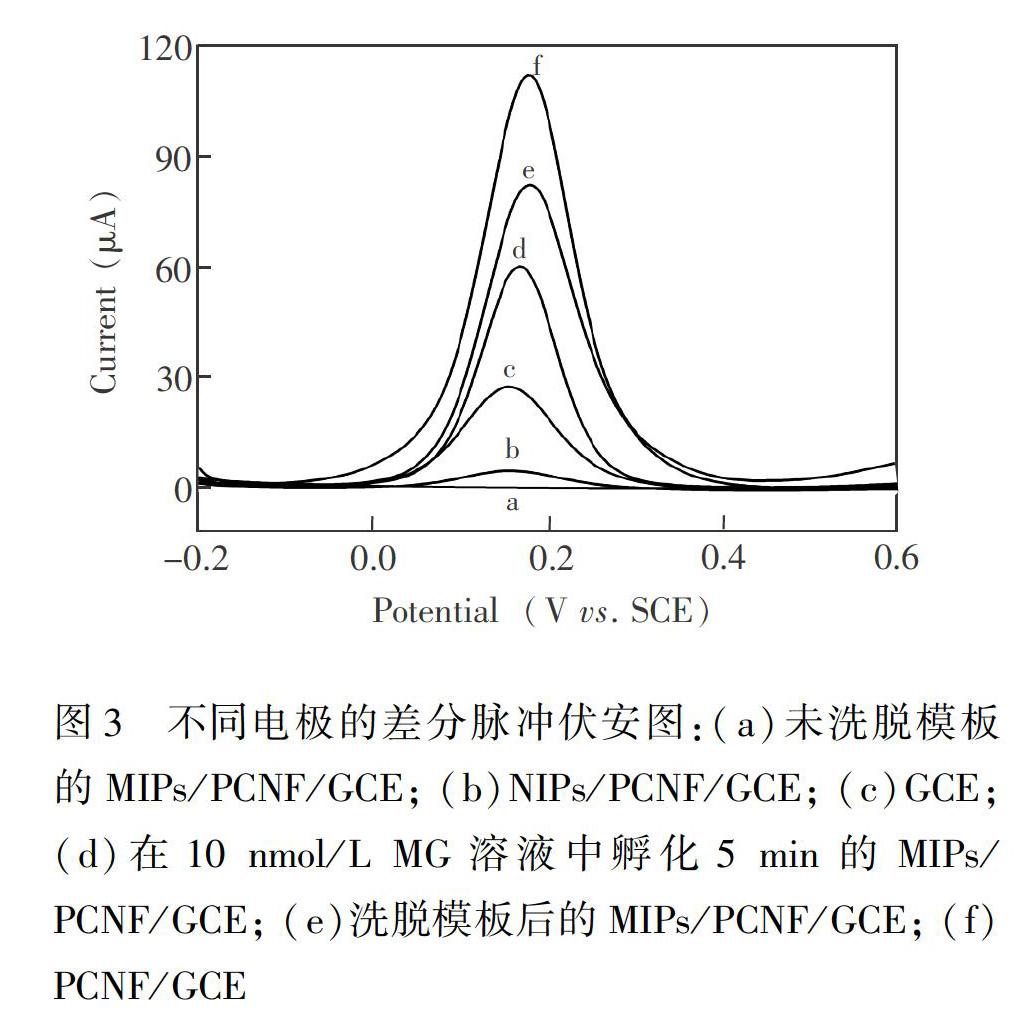
3.3.1 差分脉冲伏安法表征 图3为不同电极在含有0.10 mol/L KCl的5.0 mmol/L K3[Fe(CN)6\]溶液的DPV曲线。由图可见,PCNF/GCE(曲线f)的[Fe(CN)6\]3-4电流远高于裸玻碳电极(曲线c),这是由于修饰的PCNF材料提高了电极的导电性及活性表面积; PCNF/GCE表面电聚合了分子印迹膜后,无明显电流峰(曲线a),说明修饰电极表面已被致密的非导电聚合膜覆盖,阻碍了[Fe(CN)6\]3-4向电极表面扩散; 洗脱模板分子后的印迹电极出现了一个较大的电流峰(曲线e),是因为分子印迹膜形成大量的印迹孔穴, 使[Fe(CN)6\]3-4可以到达电极表面发生氧化还原反应而产生电流信号; 将洗脱模板分子的印迹电极重新吸附MG,其峰电流明显下降(曲线d),原因是MG占据了部分印迹孔穴,导致印迹电极表面的[Fe(CN)6\]3-4扩散通道减少。非印迹电极(曲线b)由于NIPs膜的非导电特性和无印迹位点而产生很弱的DPV电流响应。
3.3.2 电化学阻抗表征 电化学阻抗是表征电极表面电子转移动力学的一种有效方法。在EIS谱图中,高频区半圆的直径等于电子转移电阻Rct。图4为不同电极的电化学阻抗图谱,可见裸电极具有较小的电子转移电阻(337 Ω,曲線b),而PCNF/GCE的电子转移电阻显著减小(31 Ω,曲线a),说明PCNF的引入促进了电极与电解质之间的电子传递; 非印迹电极(4473 Ω,曲线f)和未洗脱模板(5214 Ω,曲线e)的印迹电极由于表面覆盖了非导电聚合膜,阻碍了[Fe(CN)6\]3-4的电子传递,导致电子转移电阻急剧增大; 洗脱模板分子后,印迹膜表面形成大量印迹孔穴,有利于[Fe(CN)6\]3-4的渗透和电子转移,印迹电极的电子转移电阻显著减小(960 Ω,曲线c); 重新吸附MG后的印迹电极的电子转移电阻明显增加(1956 Ω,曲线d),说明一部分印迹孔穴又被堵塞,致使电子传递受阻,与DPV表征结果相符。
3.3.3 循环伏安法表征 在含有0.10 mol/L KCl的5.0 mmol/L K3[Fe(CN)6\]溶液中,洗脱模板后的MIPs/PCNF/GCE的循环伏安曲线见图5,可见随着扫描速率增加,峰电流逐渐增大。阳极峰电流(ipa)和阴极峰电流(ipc)与扫描速率的平方根呈良好的线性关系,线性方程分别为ipa=5.828v1/2+6.815(R2=0.9989)和ipc=-6.511v1/2-14.074 (R2=0.9996), 说明[Fe(CN)6\]3-4在MIPs/PCNF/GCE上的电化学反应是一个扩散控制过程[22]。
3.4 实验条件的优化
3.4.1 PCNF涂覆量的影响 考察了玻碳电极上修饰不同用量(0、2.0、4.0、6.0、8.0和10.0 μL)PCNF的响应电流变化,结果表明,当PCNF涂覆量为4.0 μL时,PCNF修饰电极上的峰电流最大,即4.0 μL为PCNF的最佳用量。
3.4.2 模板分子与功能单体摩尔比的影响
传感器制备过程中模板分子与单体的摩尔比影响印迹膜中印迹位点的数量,进一步影响传感器的再结合和识别性能。在含有固定浓度MG(0.10 mmol/L)和不同浓度邻苯二胺(摩尔比为1∶1、1∶2、1∶4、1∶6、1∶10、1∶20和1∶40)的电聚合溶液中制备了不同的传感器。结果表明,模板分子与单体摩尔比为1∶4的传感器具有最大的响应电流。当摩尔比大于1∶4时,响应电流较小,这是因为单体的数量太少,不能结合足够的模板分子。而过量的单体可能占据了电极表面,使得有效的识别位点减少,因而响应电流减小。因此,模板分子与单体的最佳摩尔比为1∶4。
3.4.3 扫描圈数的影响 电聚合扫描圈数会影响聚合膜的厚度,从而影响传感器的灵敏度和稳定性。考察了不同聚合圈数的电极的性能。结果显示,当扫描圈数从5圈增加到20圈时,峰电流显著增加。当扫描圈数超过20圈时,峰电流随扫描圈数的增加而减小,可能是形成较厚的聚合膜,导致位于聚合物膜中心区域的模板分子不能完全去除。选择20圈作为最佳扫描圈数。
3.4.4 洗脱剂和洗脱时间的影响 模板分子洗脱直接影响印迹孔穴的数量,所选洗脱剂应尽可能将模板分子洗脱下来而不破坏印迹聚合物膜的结构。基于孔雀石绿易溶于水、乙醇和甲醇,而酸能够破坏模板分子与功能单体之间结合的氢键,选择纯水、无水乙醇、纯甲醇、10%(V/V) HAc溶液、10%(V/V) HAc-乙醇溶液、10%(V/V) HAc-甲醇溶液、1.7 mol/L HCl溶液为洗脱剂,考察洗脱模板分子后的印迹电极的电流响应。结果发现,10%(V/V) HAc-甲醇溶液和10%(V/V) HAc-乙醇溶液洗脱效果最好,其次是10%(V/V) HAc溶液、纯甲醇、无水乙醇,再次是1.7 mol/L HCl,而纯水的洗脱效果最差。由于10%(V/V) HAc-甲醇溶液稍优于10%(V/V) HAc-乙醇溶液,因此选择其为洗脱剂进一步优化其配比。考察10%,20%、30%、40%、50% (V/V)的HAc-甲醇溶液洗脱模板分子的效果。结果发现,以20%(V/V) HAc-甲醇为洗脱液,印迹电极孵化前后的电流变化值最大,随HAc体积分数增大,电流变化值下降,可能是HAc与MG之间极易形成氢键,有利于模板分子洗脱,但HAc的用量过大可能破坏印迹膜的结构,使得印迹膜的印迹效果变差。故选择20%(V/V) HAc-甲醇溶液为洗脱液。同时考察印迹电极在20%(V/V) HAc-甲醇洗脱液中洗脱不同时间的效果。研究发现,随着洗脱时间增加,峰电流不断增大,当洗脱时间超过25 min时,峰电流不再增大,这说明洗脱时间太短,印迹膜上的模板分子未能完全去除; 洗脱时间达到25 min时,模板分子已完全被洗脱下来; 继续延长洗脱时间,不仅影响实验效率,还可能破坏印迹聚合物膜的结构。因此,选择洗脱时间为25 min。
3.4.5 样品溶液pH值的影响 以含10.0 nmol/L MG的0.20 mol/L磷酸盐溶液为样品溶液,考察印迹电极在pH 4.5~8.5的样品溶液中孵化对孔雀石绿的选择吸附效果。结果表明, 在pH 7.0时峰电流降至最低,说明在中性溶液中吸附效果最好。这可能因为MG的pKa=6.9,当样品溶液pH
3.4.6 孵化时间的影响 将洗脱模板后的印迹电极置于10.0 nmol/L MG溶液中,对孵化时间进行优化。结果显示,峰电流随孵化时间的延长而减小,20 min后峰电流趋于平稳,说明模板分子与印迹电极之间达到了吸附平衡。因此,选择孵化时间为20 min。
3.5 分子印迹传感器的分析性能
3.5.1 线性范围及检出限 在最优条件下,利用MIPs/PCNF/GCE对不同浓度的MG進行测试。如图6所示, DPV峰电流随MG浓度的增加而逐渐减小,在0.10~10.0 nmol/L浓度范围内,峰电流(μA)与MG浓度(nmol/L)呈良好的线性关系(图6插图),线性方程为i(μA)=3.672c(nmol/L) + 79.39(R2=0.997),检出限为0.042 nmol/L(3S/k),其中S为空白溶液测定8次的峰电流的标准偏差,k为校准曲线斜率的绝对值。本方法的检出限低于大多数文献报道的电化学方法(表1),高于文献报道的Ru(bpy)2+3电化学发光法[16],但其使用的发光试剂Ru(bpy)2+3价格昂贵。
3.5.2 选择性及干扰性 为了评价传感器的选择性,选择结晶紫、氯霉素、洛美沙星等几种常见的抗菌药物作为干扰物质。定义选择因子[26]K=△i1/△i,
其中, △i1为空白电极产生的峰电流与存在干扰物质产生的峰电流的差值,△i为空白电极产生的峰电流与存在MG产生的峰电流差值,K值越小,证明印迹电极对模板分子的选择性越好。将洗脱模板分子后的印迹电极分别在10.0 nmol/L的结晶紫、氯霉素、洛美沙星中吸附5 min后,进行DPV扫描。结晶紫、氯霉素、洛美沙星的选择因子分别为0.47、0.22和0.15。尽管结晶紫与MG的结构相似度高,使得印迹膜上的孔穴对结晶紫产生吸附,产生一定的电流响应,但未对MG的测定形成明显干扰,表明印迹膜与MG的结合能力除了两者互补的官能团决定外,印迹膜上的孔穴的空间结构对两者的结合作用也产生较大的影响。结果表明,此传感器能够对MG进行特异性识别。同时也考察NIPs/PCNF/GCE对结晶紫、氯霉素、洛美沙星的电流响应。发现NIPs传感器对上述物质响应不灵敏,对MG无特异性识别作用。
此外,在10.0 nmol/L MG溶液中,考察了一些可能共存的生物小分子和离子对MG测定的影响。结果表明,200倍的赖氨酸和葡萄糖,500倍的Ca2+、Al3+、Mg2+、Na+、SO2-4、Cl、PO3-4对于MG的测定无显著影响(电流信号变化小于5%),表明所构建的传感器具有良好的抗干扰能力。
3.5.3 重現性和稳定性 在相同条件下制备的5支印迹电极分别对2.0 nmol/L MG溶液进行测定,测得峰电流的相对偏差(RSD)为7.2%。同一支印迹电极平行测定6次,RSD为3.6%。将5支印迹电极保存于4℃下,10天后再对2.0 nmol/L MG溶液测定,电流响应为初始响应值的91%,表明此电极有良好的重现性和稳定性。
3.6 实际样品分析
将MIPs/PCNF/GCE用于市售新鲜草鱼及养殖水样中的MG检测,草鱼及养殖水样中未检出MG。对实际样品进行了加标回收实验。由表2可知,不同加标浓度下的回收率为82.0%~96.5%, RSD<8.0%,说明此方法对于不同基质样品中MG的测定具有较高的准确度和精密度,可用于实际样品中MG的检测。
4 结 论
构建了一种基于PCNF的MG分子印迹电化学传感器。在0.10~10.0 nmol/L浓度范围内,MIPs/PCNF/GCE对MG表现出良好的线性响应,检出限为0.042 nmol/L,低于大多数文献报道的电化学方法。此传感器不仅对MG具有较高的灵敏度和选择性,而且具有良好的重现性和稳定性,为MG提供了一种简单、经济、灵敏的电化学检测方法。将此传感器用于检测市售新鲜草鱼及养殖水样品中MG含量,结果令人满意,有望应用于实际样品中MG残留量的检测。
References
1 LIU Ming-Yang, XIAO Shan-Shan, YU Bing, YANG Chun-Guang, ZHAO Jing-Hong, JIN Yan, ZHU Cheng-Yun. Journal of Food Safety & Quality, 2015, 6(1): 35-40
刘名扬, 肖珊珊, 于 兵, 杨春光, 赵景红, 金 雁, 朱程云. 食品安全质量检测学报, 2015, 6(1): 35-40
2 Bueno M J M, Herrera S, Uclés A, Agüera A, Hernando M D, Shimelis O, Rudolfsson M, Fernández-Alba A R. Anal. Chim. Acta, 2010, 665(1): 47-54
3 Srivastava S, Sinha R, Roy D. Aquat. Toxicol., 2004, 66(3): 319-329
4 Li L, Peng A H, Lin Z Z, Zhong H P, Chen X M, Huang Z Y. Food Chem., 2017, 229: 403-408
5 Long C, Mai Z, Yang Y, Zhu B,Xu X, Lu L, Zou X. J. Chromatogr. A, 2009, 1216(12): 2275-2281
6 WANG Li-Bo, SU Li-Qiang, HAN Chao-Nan, L Yan-Rong. Journal of Analytical Science, 2018, 34(2): 229-233
王丽博, 苏立强, 韩超男, 吕艳荣. 分析科学学报, 2018, 34(2): 229-233
7 GONG Xiao-Ming, HUA Meng-Meng, WANG Hong-Tao, WANG Bing-Jun, MA Rong-Hui. Journal of Instrumental Analysis, 2017, 36(7): 897-901
宫小明, 华萌萌, 王洪涛, 王炳军, 马荣桧. 分析测试学报, 2017, 36(7): 897-901
8 Kwan P P, Banerjee S, Shariff M, Ishak N A S, Yusoff F M. Food Control, 2018, 92: 101-106
9 Maxwell E J, Tong W G. J. Chromatogr. B, 2016, 1020: 29-35
10 Xu K X, Guo M H, Huang Y P, Li X D, Sun J J. Talanta, 2018, 180: 383-388
11 Zhang K, Song G, Yang L, Zhou J, Ye B. Anal. Methods, 2012, 4(12): 4257-4263
12 Yi H, Qu W, Huang W. Microchim. Acta, 2008, 160(1-2): 291-296
13 TANXue-Cai, WU Jia-Wen, HU Qi, LI Xiao-Yu, LI Peng-Fei, YU Hui-Cheng, LI Xiao-Yan, LEI Fu-Hou. Chinese J. Anal. Chem., 2015, 43(3): 387-393
谭学才, 吴佳雯, 胡 琪, 李晓宇, 李鹏飞, 余会成, 李小燕, 雷福厚. 分析化学, 2015, 43(3): 387-393
14 Lu B, Xia J, Wang Z, Zhang F, Yang M, Li Y, Xia Y. RSC Adv., 2015, 5: 82930-82935
15 LI Ying, XU Wen-Kai, LI Ping, ZHU Xiao-Xue, HUANG Yan-Feng, ZHANG Ji-Mei. Chinese J. Anal. Chem., 2018, 46(7): 1047-1054
李 穎, 徐文凯, 李 苹, 朱小雪, 黄艳凤, 张纪梅. 分析化学, 2018, 46(7): 1047-1054
16 Huang B, Zhou X, Chen J, Wu G, Lu X. Talanta, 2015, 42: 228-234
17 Yang B, Fu C, Li J, Xu G. TrAC-Trend. Anal. Chem., 2018, 105: 52-67
18 Sun J, Liu Y, Lv S, Huang Z, Cui L, Wu T. Electroanalysis, 2016, 28(3): 439-444
19 Sakthivel M, Ramaraj S, Chen S M, Dinesh B, Ramasamy H V, Lee Y S. Anal. Chim. Acta, 2018, 1006: 22-32
20 Qin D, Gao S, Wang L, Shen H, Yalikun N, Sukhrobov P, Wagberg T, Zhao Y, Mamat X, Hu G. Microchim. Acta, 2017, 184: 2759-2766
21 Yuan D, Huang X, Yan J, Yu W, Meng H, Rong J. Sci. Adv. Mater., 2013, 5(11): 1-7
22 Han S, Li B, Song Z, Pan S, Zhang Z, Yao H, Zhu S, Xu G. Analyst, 2017, 142(1): 218-223
23 GAI Pan-Pan, GUO Zhi-Yong, DUAN Jing, ZHANG Hui-Na, WANG Sui, WEI Dan-Yi. Chinese Journal of Sensors and Actuators, 2010, 23(8): 1062-1065
盖盼盼, 郭智勇, 段 静, 张会娜, 王 邃, 魏丹毅. 传感技术学报, 2010, 23(8): 1062-1065
24 Huang W, Yang C, Qu W, Zhang S. J. Electrochem., 2008, 44(8): 946-951
25 Zhou Y, Li X, Pan Z, Ye B, Xu M. Food Anal. Methods, 2019, 12(5): 1246-1256
26 LUAN Chong-Lin, LI Ming-Jie, LI Zhong-Jin, SU Xiu-Xia, JIANG Xiao-Hua. Physical Testing and Chemical Analysis Part B: Chemical Analysis, 2011, 47: 885-893
奕崇林, 李铭杰, 李仲谨, 苏秀霞, 蒋晓华. 理化检验-化学分册, 2011, 47: 885-893
Abstract A molecularly imprinted electrochemical sensor for specific recognition of malachite green (MG) was prepared by electropolymerization of o-phenylenediamine on the surface of porous carbon nanofiber (PCNF) modified glassy carbon electrode with MG as template molecule. The electrochemical performance of the sensor was characterized by differential pulse voltammetry (DPV), cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS). The experimental parameters, such as the amount of PCNF, molar ratio of template molecule to functional monomer, scan cycle of CV, eluent, elution time, pH value of sample solution and incubation time, were optimized. Under optimal conditions, the peak current was linear to MG concentration in the range of 0.10-10.0 nmol/L, with a detection limit of 0.042 nmol/L. The prepared sensor possessed excellent selectivity, reproducibility and stability, and was applied to the detection of MG in commercially available fresh grass carp and aquaculture water samples with recoveries of 82.0%-96.5%.
Keywords Molecularly imprinted electrochemical sensor; Electropolymerization; Porous carbon nanofiber; Malachite green
