
马来酸酐接枝改性低密度聚乙烯的制备及其性能研究
2020-03-03 06:11叶翠翠丁小军李林繁李景烨
辐射研究与辐射工艺学报 2020年1期
叶翠翠 丁小军 李林繁 李景烨,3
1(中国科学院上海应用物理研究所 上海 201800)
2(中国科学院大学 北京 100049)
3(上海师范大学 上海 200234)
低密度聚乙烯(LDPE)具有密度小、易加工、耐化学腐蚀、机械性能良好以及价格低廉等优点,在包装材料、通用电缆以及薄膜材料等领域都有着广泛的应用[1-4]。然而,LDPE的熔点低、尺寸稳定性差以及耐热性差等缺点,使其在实际应用中受到一定的限制。通过与其他聚合物熔融共混,可制得综合性能优异的LDPE复合材料。常用来共混改性LDPE 的聚合物有尼龙6(PA6)[5-7]、聚偏氟乙烯(PVDF)[8-9]和聚对苯二甲酸乙二醇酯(PET)[10]等,但是LDPE 与这些聚合物之间的相容性较差,共混体系的物理性能相对较差。通常来说,在不相容共混物中加入适当的增容剂是提高共混物组分间相容性的有效方法。增容的方法主要包括物理增容和化学增容两种。物理增容是选用合适的嵌段共聚物或者接枝共聚物,利用其链段分别与共混物中两组分相同或者相容的特性以达到增容的效果[11-12]。然而,无论是在学术还是工业角度上,为每一对不相容共混体系设计以及合成特定的增容剂都是非常困难的。化学增容是利用挤出或共混过程中,反应性基团之间的相互反应原位合成增容剂[13-15]。由于基团之间的反应往往发生在界面处,生成的增容剂可以稳定地处于不相容共混物的两相界面而达到增容的效果,然而,化学增容的方式要求聚合物上存在特殊的反应性基团,可以在共混或挤出的短时间内充分反应。因此,对于LDPE等不含反应性基团的非极性聚合物进行功能性单体接枝改性具有十分重要的意义。
马来酸酐(MAH)是一种含有多官能团的小分子单体,其双键可以在引发剂或者辐照的条件下与聚合物发生聚合反应,酸酐基团可以与氨基、羟基以及羧基等基团发生反应[16]。由于MAH单体的对称结构,MAH 的自聚较为困难,在接枝的过程中不会在LDPE上形成长支链结构,从而避免了LDPE 机械性能的急剧下降[17]。目前,通过MAH接枝改性LDPE在国内外已经有诸多研究,主要的方法有溶液接枝法[18]、熔融接枝法[19-21]和固相接枝法[22]等。He 等[18]以二甲苯为溶剂,过氧化二异丙苯(DCP)为引发剂,通过溶液法将MAH 单体接枝到了LDPE的分子链上,结果表明MAH 的接枝率与DCP 的质量分数有关。在最佳实验条件下,样品的MAH 接枝率可达4%。溶液接枝法可以得到接枝率较高的产物,但是样品的后续处理较为复杂。因此,更多的研究者采取熔融接枝的方式来接枝改性LDPE。Iqbal 等[20]通过双螺杆挤出的方式制备了LDPE-g-MAH,通过红外表征确定MAH 成功地接枝到LDPE 的分子链上,且接枝率与力学性能随着引发剂过氧化二苯甲酰(BPO)质量分数的增加呈先增加后降低的趋势。同时,制备得到的LDPE-g-MAH 作为增容剂显著地提高了LDPE 与PA6 之间的相容性,为LDPE 的共混改性提供了更多的潜在可能。然而熔融接枝的过程中,LDPE会发生交联等副反应,接枝率较低,且MAH 的熔点较低,在熔融共混或者挤出时容易挥发,对反应熔腔以及人体呼吸道有一定的危害性。相较于溶液接枝法和熔融接枝法,固相辐射接枝具有操作简单,反应温度低,产物较为纯净等优点。Chaudhari等[23]通过固相辐射接枝的方式将甲基丙烯酸(MAA)接枝到LDPE 的薄膜表面,结果表明MAA的接枝率可以通过吸收剂量、单体浓度、抑制剂浓度以及薄膜厚度等因素进行调控,同时发现该改性薄膜在水相介质中对染料分子以及重金属离子具有良好的吸附能力。然而,表面接枝改性的样品,样品内部的结构和性能并未发生改变,功能化改性仅存在于样品的表面,在一定程度上限制了改性样品的应用范围。此外,尽管有大量关于辐射接枝的小分子单体改性LDPE研究,但是MAH辐射接枝LDPE的研究还鲜有报道。
我们首先通过简单的熔融共混的方式,制备得到LDPE/MAH 二元共混物,而后选用室温辐照的方式将MAH 单体成功地接枝到LDPE 的分子链上,并通过傅里叶红外光谱(FTIR)表征了LDPE-g-MAH的化学结构。选用二甲苯作为溶剂,以索氏抽提的方式测定了不同吸收剂量下样品的凝胶质量分数。通过示差扫描量热法(DSC)、热重分析法(TGA)和X 射线衍射谱(XRD)等研究了吸收剂量对样品热稳定性能和结晶性能的影响,并测定了制备所得的一系列LDPE-g-MAH 样品的力学性能。
1 材料与方法
1.1 原料和试剂
LDPE,2426H,熔融指数为1.9 g/10 min,密度为0.9 g/cm3,购于中国石化集团茂名石油化工有限公司;MAH,购于上海展云化工有限公司;丙酮(分析纯),杭州双林化工试剂有限公司。
1.2 仪器和设备
多功能精密混炼机,Haake Polylab QC,赛默飞世尔有限公司,美国;傅里叶变换红外光谱仪,Bruker VERTEX 70V,布鲁克公司,德国;示差扫描量热仪(DSC),Q2000,TA 仪器有限公司,美国;热重分析仪,Q500,TA 仪器有限公司,美国;扫描电子显微镜,Hitachi S-4800,日立公司,日本;X 射线衍射仪,Bruker-D8,布鲁克仪器公司,德国;万能拉伸试验机,Instron-5966,英斯特朗公司,美国。
1.3 LDPE/MAH 二元共混物的制备
熔融共混前,先将LDPE 置于80°C 的真空烘箱中恒温干燥8 h 以去除水分,随后将LDPE 与MAH 以99/1、98/2、95/5、92/8、90/10 的质量比经简单物理混合后加入多功能精密混炼机。所有样品首先在160°C 下20 r/min 熔融共混2 min,随后将转速提升至50 r/min 再熔融共混5 min,制备得到一系列MAH 含量不同的LDPE/MAH 二元共混物,分别标记为LDPE/MAH((100−X)/X)。其中,X为MAH的添加量。然后将LDPE/MAH的二元共混物热压成厚度为0.3 mm 的薄膜。具体方法:先将一定质量的样品置于厚度为0.3 mm 的模具当中,在160°C下热熔5 min使其充分熔融,随后在10 MPa的压力下恒温保压2 min,最后在相同的压力下冷压2 min,制备得到LDPE/MAH 的共混薄膜。
1.4 LDPE-g-MAH的辐射制备
将1.3 节中制备得到的不同MAH 质量分数的LDPE/MAH 二元共混物薄膜干燥后置于聚乙烯的自封袋中,用真空自动封口机密封后,在室温下进行钴源辐照。吸收剂量为10 kGy、25 kGy、50 kGy和100 kGy,γ 射线辐照时间均为17 h。将制备得到的辐照样品置于索氏抽提设备中,用丙酮溶液抽提48 h 以去除未反应的MAH 单体及MAH 的低聚物。将样品彻底干燥后得到纯净的LDPE-g-MAH。
1.5 表征与分析方法
1.5.1 凝胶质量分数的测定
通过索氏抽提的方式测定辐照制备得到的LDPE-g-MAH 样品的凝胶质量分数。将称重好的LDPE-g-MAH 样品置于邻二甲苯溶液中抽提36 h,再将抽提以后的样品置于鼓风烘箱中干燥至恒重,精准称量干燥后LDPE-g-MAH 样品的质量,最后将抽提前后样品的质量按式(1)进行计算。抽提进行3次平行实验,取平均值为最终的实验结果。

式中:fgel是最终样品凝胶的质量分数;m1与m2则为抽提前后LDPE-g-MAH样品的质量(g)。
1.5.2 FTIR分析
通过FTIR 对样品进行表征。将待测样品通过平板硫化机热压成超薄膜后,采用透过模式进行测试。测试的波数范围为4 000~400 cm−1,在分辨率为2 cm−1下累积扫描64次。
1.5.3 热性能分析
通过DSC 表征了所有样品的结晶以及熔融行为。称取5~6 mg 的样品置于铝坩埚中,在氮气氛围下进行测试。首先以10°C/min 的速率将样品从30°C升温至180°C,恒温5 min以消除热历史,随后以10°C/min的降温速率降温至30°C,记录其结晶行为,最后再以相同的升温速率将样品升温至180°C,记录其熔融行为。
通过TGA 表征样品的热稳定性能。在氮气氛围下,所有待测样品从30°C升温至650°C,升温速率为10°C/min,记录样品的热降解行为。
1.5.4 形貌表征
通过SEM 观察样品的断面形貌。首先将样品置于液氮中进行淬断,在55°C的真空烘箱干燥3 h后进行喷金处理,加速电压为3 kV。
1.5.5 晶体以及片晶结构表征
通过XRD表征了样品的晶型。测试范围为5°~45°,扫描速率为1(°)/min。
1.5.6 力学性能表征
通过万能试验拉伸机测试了样品的力学性能。测试样条均为标准哑铃型,拉伸速率为10 mm/min,样品的夹持距离为18 mm。所有样品均进行5次以上的平行测试,取其平均值作为最终结果。
2 结果与讨论
2.1 LDPE/MAH二元共混物形貌
图1 是不同质量配比的LDPE/MAH 二元共混物断面的SEM图。共混物中,MAH的质量分数分别为1%、2%、5%、8%和10%。从图1 中可以发现,当MAH 的质量分数少于2%时,共混物中没有发生明显的相分离,呈均相结构;当MAH的质量分数在5%~10%时,共混物中出现了明显的相区。这表明当MAH 质量分数较高时,MAH 单体在LDPE 的基体中发生了团聚。后续实验选用MAH 质量分数为1%和2%的LDPE/MAH 的二元共混物进行研究。

图1 不同质量配比的LDPE/MAH二元共混物断面的SEM图Fig.1 SEM images of LDPE/MAH blends with different mass ratios
2.2 LDPE/MAH二元共混物的辐射效应
2.2.1 凝胶质量分数
LDPE是典型的辐射交联聚合物。据相关文献报导[24-26],辐照过程中,LDPE在相对较低吸收剂量下(10 kGy~200 kGy)以辐射交联反应为主,随着吸收剂量进一步上升,辐射降解反应逐步加剧,从而占主导地位。因此,本工作中将纯LDPE以及不同配比的LDPE/MAH 二元共混物在不同的吸收剂量(0 kGy、10 kGy、25 kGy、50 kGy、100 kGy)以及室温真空氛围下进行辐照。此外,通过索氏抽提的方式研究了LDPE/MAH 二元共混物在不同吸收剂量下的交联程度,选用的溶剂为二甲苯,因为LDPE可以在二甲苯中溶解,而交联的LDPE在二甲苯中不能溶解只能发生溶胀。
图2是纯的LDPE以及LDPE/MAH二元共混物在不同吸收剂量下的凝胶质量分数。如图2 所示,随着吸收剂量的上升,所有样品的凝胶质量分数均逐渐增加。这是由于随着吸收剂量的增加,样品中产生的自由基数量也在逐步上升,从而增加了自由基之间的有效碰撞概率[27],促使交联网络结构的逐步形成,最终提高了被照样品的凝胶质量分数。从图2 中还可以发现,在同一吸收剂量下,LDPE/MAH 共混物的凝胶质量分数在整个研究范围内均要低于纯的LDPE,这是由于随着吸收剂量的增加,LDPE 的大分子自由基与MAH 单体的反应概率也随之上升,LDPE 与MAH 之间发生接枝反应,在一定程度上减少了LDPE自由基之间的有效碰撞,抑制了交联网络的形成。

图2 LDPE以及LDPE/MAH共混物在不同吸收剂量下的凝胶质量分数Fig.2 Gel mass fractions of neat LDPE and LDPE/MAH blends at different absorbed doses
2.2.2 分子结构研究
为了进一步研究LDPE/MAH 二元共混物的辐射效应,我们对不同吸收剂量下辐照的LDPE以及LDPE/MAH (98/2)二元共混物进行FTIR 表征,如图3所示。图3(a)中,在1 470 cm−1以及720 cm−1处观察到的吸收峰,分别对应于纯的LDPE上亚甲基(CH2)的面内弯曲振动和主碳链上亚甲基-(CH2)n-(n>4)的面外弯曲振动[28]。此外,真空氛围下,室温辐照的LDPE 在1 741 cm−1处出现了新的吸收峰,对应于羰基(C=O)的伸缩振动峰。根据文献[29]报导,LDPE在辐照条件下会产生酮与酸酐类氧化产物。与纯LDPE的辐照样品相比,LDPE/MAH(98/2)共混物在不同吸收剂量下辐照后,除了在1 741 cm−1处出现了新的吸收峰外,在1 787 cm−1处同样可以观察到新的吸收峰,对应于MAH 上羰基(C=O)的不对称伸缩振动,如图3(b)所示。随着吸收剂量的增加,羰基(C=O)吸收峰的强度逐渐增加;当吸收剂量为100 kGy时,吸收峰的强度略微降低。这些结果表明MAH 成功地接枝到了LDPE 的分子链上,且MAH接枝率随吸收剂量的上升而上升。
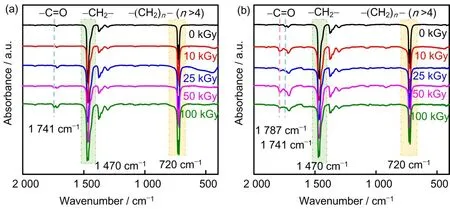
图3 纯LDPE(a)和LDPE/MAH(98/2)(b)辐照样品的FTIR图Fig.3 FTIR spectra of neat LDPE(a)and LDPE/MAH(98/2)blends(b)after irradiation
2.3 吸收剂量对LDPE/MAH 二元共混物热性能的影响
2.3.1 结晶与熔融行为
图4 为不同吸收剂量下辐照的LDPE/MAH 二元共混物(以MAH 质量分数为1%为例)的DSC曲线。如图4(a)所示,被照样品的熔点(Tm)随着吸收剂量的上升而逐渐下降。当吸收剂量较高(100 kGy)时,样品的Tm下降尤为明显。由于在辐照条件下,LDPE的晶区和非晶区均可以产生自由基,且相互之间反应的概率较低,还可以发生交联、夺氢等副反应[30]。尤其是LDPE 的辐射交联反应将会对其Tm产生显著影响,随着吸收剂量的不断上升,被照样品的交联度也随之上升,相应的溶胶部分的比例逐渐降低,而DSC 是测量溶胶部分的LDPE,因此,被照样品的Tm随吸收剂量的上升而逐渐下降。

图4 不同吸收剂量下辐照样品的DSC曲线:(a)第一次升温曲线;(b)第一次降温曲线Fig.4 DSC curves of irradiation samples at different absorbed doses:(a)the first heating curves;(b)the first cooling curves
从图4(b)可知,被照样品的结晶温度(Tc)同样随着吸收剂量的上升而逐渐下降,这是由于随着吸收剂量的上升,样品的交联程度不断上升,逐渐形成了交联网络,限制了LDPE分子链在熔体状态下的自由运动,抑制了样品的结晶能力。
2.3.2 热稳定性
图5 为不同吸收剂量下LDPE/MAH 共混物(以LDPE/MAH(98/2)为例)的热失重曲线。从图5 中可以发现,所有样品的最大热降解温度Tmax都在485°C 左右,随着吸收剂量的增加,被照样品的Tmax均向低温方向移动。这是由于LDPE 在辐照过程中会形成部分“陷落”的自由基,在高温的情况下,会加剧样品的热降解过程,导致样品热稳定性能的下降。

图5 不同吸收剂量下辐照样品的热失重曲线:(a)TGA曲线;(b)DTG曲线Fig.5 TGA(a)and DTG(b)curves of irradiation samples at different absorbed doses
2.4 吸收剂量对LDPE/MAH 二元共混物微观结构的影响
为了探究辐照对样品微观形貌的影响,我们观察了LDPE/MAH(98/2)共混物样品辐照前后断面的形貌,如图6所示。样品断面的形貌在辐照前后并未发生明显的变化,表明室温固体辐照不会对LDPE/MAH 共混物的微观形貌产生显著的影响。由于LDPE 是结晶型的聚合物,降温的过程中,LDPE发生结晶,MAH被排出到LDPE的片晶或晶纤之间,因此,LDPE 辐射接枝MAH 的反应仅存在于LDPE 的无定型区域。同时LDPE 的晶区在辐照的条件下虽然形成了“陷落”的自由基,但这些缺陷的存在并不会引起LDPE凝聚态结构的变化[31]。

图6 辐照前(a)和辐照(100 kGy)后(b)LDPE/MAH(98/2)共混物的SEM图Fig.6 SEM images of LDPE/MAH(98/2)blend before(a)and after(b)irradiation at 100 kGy
2.5 吸收剂量对LDPE/MAH 二元共混物晶体结构的影响
不同吸收剂量下LDPE-g-MAH 样品的XRD 图谱如图7所示。从图7中可以发现,未辐照的样品在21.3°和23.4°处有两个明显的吸收峰,分别对应LDPE(110)和(200)的晶面[18]。对比辐照前后样品的衍射结果,发现辐照后样品的晶型并未发生明显变化,这是由于构象的转变往往需要借助外部条件,如高温拉伸,无机填料作为长模板或者溶液中的超声作用等。本工作是在室温固态下进行辐照,LDPE的晶型不会发生明显变化。
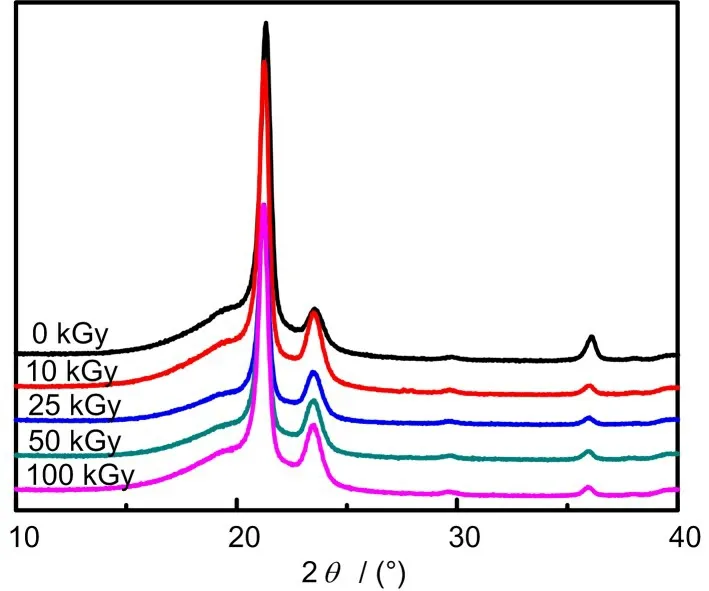
图7 不同吸收剂量下LDPE-g-MAH的XRD曲线Fig.7 XRD curves of LDPE-g-MAH at different absorbed doses
2.6 吸收剂量对LDPE/MAH 二元共混物力学性能的影响
对不同吸收剂量下辐照样品进行了拉伸性能的测试,其应力‒断裂伸长率曲线如图8 所示。从图8 中可以发现,所有样品的断裂伸长率都很高,均表现出良好的延展性,而且辐照后样品的断裂伸长率均要高于未辐照样品。此外,随着吸收剂量的上升,样品的断裂伸长率呈先增长后降低的趋势。有研究表明[32],当吸收剂量较低时,相对较低程度的交联可以提高样品的韧性;当吸收剂量较高时,样品的交联程度逐渐上升,分子链之间会形成更完善的网络结构。因此,在拉伸的过程中,分子链的运动受到限制,不易发生分子间滑移,从而导致了断裂伸长率的降低。
3 总结
本工作通过室温辐照的方式成功制备了LDPE-g-MAH 样品,详细研究了吸收剂量对LDPE-g-MAH的化学结构、微观形貌、晶体结构、热性能以及力学性能的影响。在我们的研究范围内(0 kGy~100 kGy),随着吸收剂量的上升,样品的凝胶质量分数逐渐上升,表明LDPE/MAH 共混物在辐照条件下发生了辐射交联反应。从FTIR的结果中可知,MAH 成功地接枝到了LDPE 的分子链上,且接枝率随着吸收剂量的增加而增加。此外,固体室温辐照并未改变样品的晶型以及微观形貌,但在一定程度上抑制了样品的结晶能力,这是由于被照样品在辐照的条件下发生了辐射交联,使样品形成了三维网络结构而限制了分子链的运动。样品的力学性能与吸收剂量有关,当吸收剂量较低时,轻度的交联提高了样品的韧性;当吸收剂量较高时,由于样品的交联度较高,形成了较为稳定的三维网络结构,抑制分子链段的运动能力和分子间滑移,样品的延展性略微降低。
猜你喜欢
现代应用物理(2022年2期)2022-08-11
医疗卫生装备(2022年4期)2022-05-17
辐射研究与辐射工艺学报(2022年2期)2022-04-29
科技视界(2022年9期)2022-04-09
纺织学报(2021年9期)2021-09-27
科技研究·理论版(2021年22期)2021-04-18
东华大学学报(自然科学版)(2020年6期)2021-01-20
建材发展导向(2020年16期)2020-09-25
疯狂英语·新阅版(2019年6期)2019-09-10
辐射研究与辐射工艺学报(2016年5期)2016-11-12
