
低纤维素酶背景里氏木霉菌株的构建和应用
2020-03-15 10:50刘杜娟黄火清苏小运
中国农业科技导报 2020年12期
刘杜娟,黄火清,苏小运
(中国农业科学院饲料研究所, 农业农村部饲料生物技术重点开放实验室, 北京 100081)
丝状真菌里氏木霉在工业中被广泛应用于生产纤维素酶,其分泌能力强。里氏木霉分泌的纤维素酶主要为纤维二糖水解酶(cellobiohydrolase, CBH)和内切葡聚糖酶(endoglucanase, EG),二者总计占总分泌蛋白的90%~95%[1-2]。由于具备强大的分泌潜能,且还能对蛋白质进行糖基化修饰,因此,里氏木霉被认为是一种优秀的用于重组蛋白质生产的表达宿主。对丝状真菌而言,内源酶大量分泌表达可能对以其为宿主进行的异源表达造成负面影响,但这为工程改造丝状真菌提高生产异源酶提供了一种可能性。例如,抑制米曲霉中内源性蛋白α-淀粉酶的表达可改善外源蛋白牛凝乳酶的分泌[3]。另有报道,在里氏木霉缺失CBHI的菌株中,重组内切葡聚糖酶Ⅰ的产量增加了约4倍[4],这意味着在里氏木霉中,通过遗传改造干扰内源酶的表达可能有利于异源基因的高水平表达。
对里氏木霉而言,干扰主要纤维素酶基因的表达是创建异源蛋白质生产平台的必经途径。因此,需要一种方法来有效地破坏该菌中目的基因表达。在里氏木霉中,通过基于同源性的DNA重组(homology dependent recombination, HDR)方法可以将目的基因用筛选标记基因替换从而破坏其表达[5],然而里氏木霉中HDR的效率很低。敲除参与非同源末端连接(non-homologous end joining, NHEJ)的ku70[6]或mus53基因[7]可以改善HDR的效率,但需事先制备获得NHEJ功能减弱的的受体菌株。CRISPR/Cas9系统(clustered regularly interspaced short palindromic repeats/CRISPR associated protein 9)是细菌Ⅱ型适应性免疫系统[8],其可以作为里氏木霉基因破坏的替代手段。在该系统中,gRNA识别特定的靶DNA序列,在识别位点形成DNA/RNA双链结构,引导内切核酸酶Cas9切割DNA。CRISPR/Cas9技术已广泛用于哺乳动物细胞、植物和微生物的基因组编辑,近年来已实现了通过胞内表达Cas9[9]和体外组装Cas9/gRNA复合物快速敲除里氏木霉特定基因[10]的方法,这为干扰主要纤维素酶基因的表达、创建异源蛋白质生产平台打下了良好的基础。RNAi(RNA inteference)可以沉默目的基因表达,是另一种有效干扰基因表达的方法。和CRISPR/Cas9系统相比,虽然RNAi其并不能将目的基因从基因组上敲除,但其优势在于不仅可以对多个基因(或多拷贝基因)同时进行操作,对必需基因(即敲除该基因后宿主菌无法存活)进行研究,而且操作简单。RNAi发挥作用的关键是在胞内生成靶序列特异的siRNA(small interfering RNA),而这可以通过构建质粒并将其转化进入细胞,通过特殊的设计使得所表达的RNA在胞内自动形成双链RNA结构,从而被胞内的DICER识别并切割成为siRNA[11]。
本实验室所保存的里氏木霉SUS5菌株(过表达内切葡聚糖酶EG2的尿嘧啶营养缺陷型菌株)基因组中整合了多个eg2的基因,且主要分泌CBH1和EG2两个纤维素酶。本研究尝试利用CRISPR/Cas9技术结合RNAi对eg2基因进行表达沉默,研究通过一步法快速获得低纤维素酶背景的菌株。在此基础上,又尝试表达了来自于NeosartoryafischeriP1 来源的 GH3 家族耐热β-葡萄糖苷酶NfBgl3A外源基因;相应的,也将该基因转化到正常表达纤维素酶的里氏木霉出发菌株中,以NfBgl3A基因为模型,对高、低纤维素酶表达背景下的外源基因的表达量进行了对比研究。
1 材料和方法
1.1 试验材料
1.1.1菌株和质粒 大肠杆菌Trans1-T1购自北京全式金生物技术有限公司,其余菌株和质粒均为本实验室构建并保存。基因工程菌里氏木霉SUS5为过表达内切葡聚糖酶EG2的尿嘧啶营养缺陷型菌株。基于pAPA质粒[12]为骨架构建含ampR-pyr4-ampR的重组质粒;重组质粒pEG2i用于对eg2基因进行干扰。质粒pRS-NfBgl3A-solo系将来源于Neosartoryafischeri的NfBgl3A基因插入到里氏木霉的cbh1启动子和终止子之间并构建于pRS424质粒中[13],用于耐热β-葡萄糖苷酶NfBgl3A基因表达。
1.1.2培养基 土豆培养基(1 L):土豆浸出物20 g、葡萄糖20 g、固体培养基(PDA)再加入琼脂20 g。
基础培养基(minimal medium, MM)(1 L): (NH4)2SO45.0 g、KH2PO415.0 g、 MgSO4·7H2O 0.6 g、CaCl2·2H2O 0.6 g、CoCl2·6H2O 0.003 7 g、FeSO4·7H2O 0.005 g、 ZnSO4·7H2O 0.001 4 g、MnSO4·H2O, 0.001 6 g、 碳源为2% 葡萄糖(体积分数),自然pH。
纤维素诱导培养基:MM培养基中将碳源替换为2%微晶纤维素Avicel,自然pH。
1.1.3主要试剂 尿嘧啶、PEG4000、山梨醇、滤纸、羧甲基纤维素(CMC-Na)和pNP-β-1,4-glucose(pNPG)、4-methylumbelliferyl-β-D-lactopyranoside(MUL)、DMSO购自Sigma公司;Taq酶和限制性内切酶(EcoRⅠ、NotⅠ和AscⅠ)购买自Fermentas公司;重组Cas9蛋白购自南京金斯瑞生物科技有限公司;2×TransStart FastPfu Fly PCR SuperMix和pEASY-Uni Seamless Cloning and Assembly Kit购自北京全式金有限公司;Fungal DNA Midi Kit真菌基因组提取试剂盒购自OMEGA公司;RNA纯化试剂盒购自北京天根公司;MEGAshortscript T7 Transcrip’s Kit购自Invitrogen公司(Carlsbad,CA);无缝拼接试剂盒(2×ClonExpress Mix (ClonExpress Ultra One Step Cloning Kit, Vazyme)购自南京诺唯赞生物科技有限公司。引物(表1)由上海美吉生物技术有限公司合成。

表1 本研究所用引物Table 1 Primers used in this study
1.1.4主要仪器 主要仪器包括T100TM普通PCR仪、凝胶成像系统和CFX-96实时荧光定量PCR仪,美国Bio-Rad公司;高速冷冻离心机,日本HIMAC公司;Synergy H1酶标仪购自美国Thermo公司。
1.2 pEG2i质粒构建
利用真菌基因组提取试剂盒提取里氏木霉SUS5基因组DNA,以此为模板,以Eg2-F/Eg2-R、Pdc1-F/Pdc1-R及Eno1-F/Eno1-R(表1)为引物,分别扩增503 bp的eg2基因片段(933~1 436 bp)及pdc1和eno1启动子(均为1 000 bp),1%琼脂糖凝胶电泳、回收备用。通过引物设计,使扩增得到的pdc1启动子及eno1启动子具有和pAPA质粒NotⅠ和AscⅠ两端短的重叠区域(15~20 bp)。PCR反应体系(50 μL):DNA模板1 μL,上下游引物各1 μL,FastPfu PCR SuperMix 25 μL,ddH2O 12 μL。扩增程序:95 ℃预变性5 min; 95 ℃ 30 s, 55 ℃ 30 s, 72 ℃ 90 s,34个循环;72 ℃ 10 min。首先使用Gibson assembly方法[14]构建含有对向启动子(pdc1和eno1两个启动子方向相对)的中间质粒载体pPdc1-Eno1:用NotⅠ和AscⅠ将pAPA质粒进行双酶切,然后利用pEASY-Uni Seamless Cloning and Assembly Kit处理该质粒与pdc1启动子及eno1启动子片段。反应体系(10 μL):2×Assembly Mix 5 μL,线性化质粒 3 μL,基因片段共1.0 μL,dd H2O 1.0 μL。转化TransI-T1并筛选阳性克隆。同样构建对eg2基因进行干扰的重组质粒pEG2i:首先将质粒pPdc1-Eno1用EcoRⅠ酶切线性化,再与上述扩增得到的eg2基因无缝拼接,热激转化TransI-T1并筛选阳性克隆。经PCR及测序验证得到正确重组的质粒pEG2i。
1.3 设计靶向chh1的gRNA
使用E-CRISPR服务器(http://www.e-crisp.org/E-CRISP/)设计针对cbh1的gRNA序列[10]。在gRNA的上游设计T7启动子(5′-TAATACGACTCACTATAGG-3′),相应的合成互补的两条单链脱氧核糖核苷酸引物gRNA-cbh1-F3和gRNA-cbh1-R3(表1)。等量(1 μg)混合两种互补寡核苷酸,沸水浴10 min,室温下缓慢冷却以退火。使用该退火的双链DNA作为模板,使用MEGAshortscript T7 Transcrip’s Kit体外转录获得gRNA,使用RNA纯化试剂盒纯化。
1.4 里氏木霉转化
将里氏木霉孢子接种于PDA平板,28 ℃静置培养,5 d后收集新鲜孢子,用灭菌棉签刮下孢子后溶于水,收集孢子悬液并通过200目筛过滤去除菌丝及杂质,接种1×107个孢子于50 mL土豆培养基中,28 ℃、160 r·min-1震荡培养12 h。用200目筛过滤收集萌发的菌丝,以无菌水及1.2 mol·L-1MgSO4溶液洗涤,之后溶于15 mL含150 mg Lysing enzymes 的1.2 mol·L-1MgSO4溶液,28 ℃消化2 h后收集原生质体。12 mg Cas9与6.5 mg gRNA混合,室温孵育30 min,得到Cas9/gRNA复合物。以PEG介导的原生质体转化法[15]将5 mg pEG2i质粒和Cas9/gRNA复合物共同转入里氏木霉SUS5原生质体,涂布于含1 mol·L-1山梨醇的MM-glucose琼脂糖培养基上生长。
4 d后随机挑取平板上的转化子,接种于含MM-glucose培养基的固体平板上,28 ℃继续静置培养3~4 d。挑取平板上菌落少许菌丝,使用UniversAll组织提取/ PCR试剂盒提取基因组DNA作为模板,以YZ-Eg2-F/R为引物(表1)PCR验证pEG2i质粒是否已整合进入里氏木霉的基因组。对Cas9/gRNA介导的cbh1基因组座位的编辑,用YZ-cbh1F/R为引物(表1)PCR验证。同上采取PEG介导的原生质体转化法将pRS-NfBgl3A-solo质粒转入里氏木霉细胞中,挑转化子验证。
1.5 纤维素酶和异源β-葡萄糖苷酶基因诱导表达
将2×107个里氏木霉新鲜孢子接种于50 mL MM-glucose种子培养基中,28 ℃、180 r·min-1振荡培养2 d。用12层纱布过滤收集菌丝,并用无菌水充分冲洗、滤干。将等量湿重(1 g)的菌丝转接于50 mL 诱导培养基中(MM-Avicel),28 ℃、180 r·min-1振摇培养。从24 h 开始收集发酵液,每24 h 收集一次,酶液样品存于4 ℃备用。
1.6 纤维素酶酶活测定
1.6.1β-葡萄糖苷酶酶活测定 用对硝基苯-β-D-葡萄糖苷(pNPG)作为底物进行测定。混合125 μL 4 mmol·L-1的pNPG溶液和275 μL的柠檬酸-磷酸氢二钠缓冲液(50 mmol·L-1,pH 5.0),50 ℃水浴平衡。加入适当稀释的酶液100 μL,50 ℃水浴保温10 min,向反应体系中加入1.5 mL Na2CO3试剂以终止反应,测定405 nm的吸光度。在50 ℃、pH 5.0的条件下,每小时水解pNPG,产生1 μmol的pNP所需要的酶量定义为一个酶活力单位(U)。
1.6.2外切纤维素酶活的测定 称取15 mg MUL溶于500 μL DMSO,再将其转移到30 mL柠檬酸缓冲液(pH 5.0,50 mmol·L-1)中。将25 μL适当稀释的酶液、200 μL MUL和25 μL葡萄糖(1 mol·L-1)混合,此为不加纤维二糖实验组。在混合溶液中,葡萄糖会抑制β-葡萄糖苷酶降解MUL,因此,测得的活性实际为外切纤维素酶活(CBH1)和内切纤维素酶酶(EG)的活性。同时添加纤维二糖实验组:25 μL酶液和200 μL MUL、25 μL葡萄糖(1 mol·L-1)、25 μL(50 mmol·L-1)纤维二糖,于50 ℃反应20 min。加入纤维二糖会抑制CBH1的活性,因此测得的是EG的活性。终止反应时,加入250 μL Na2CO3(1 mol·L-1),于370 nm测定吸光度。将不加纤维二糖实验组酶活减去添加纤维二糖实验组酶活,即为外切纤维素酶较为特异的活性。一单位外切纤维素酶酶活定义为每分钟催化1 nmol MUL水解所需要的酶量。
1.6.3内切纤维素酶酶活测定 采用羧甲基纤维素钠(CMC-Na)作为底物进行测定。用柠檬酸-磷酸氢二钠缓冲液(50 mmol·L-1、pH 5.0)配制1.5%的羧甲基纤维素钠溶液。羧甲基纤维素钠溶液应立即使用,使用前适当摇匀。取适当稀释的酶液100 μL,加入到900 μL CMC-Na底物中,振荡混合均匀,50 ℃水浴10 min,向各试管中加入1.5 mL DNS试剂,再向空白中加酶液0.1 mL,混合均匀。在沸水中煮5 min,迅速冷却,测定540 nm的吸光度。在50 ℃、pH 5.0的条件下,每小时水解羧甲基纤维素钠,产生1 μmol还原糖(以葡萄糖计)所需要的酶量定义为一个酶活力单位(U)。
1.7 RT-qPCR
接种2×107个里氏木霉孢子至MM+glucose液体培养基中,28 ℃、180 r·min1振摇培养48 h,收集菌丝。在液氮中研磨菌丝,采用TRIzol试剂盒提取总RNA,并利用HiScriptⅡ 1st Strand cDNA Synthesis Kit试剂盒将mRNA反转录得到cDNA。以actin基因作为内参,通过荧光定量PCR(RT-qPCR-EG2F/R,表1)检测eg2的转录水平,用2-ΔΔCT[16]方法对目的基因相对转录丰度进行比较。
2 结果与分析
2.1 chh1的基因敲除
将成功构建的pEG2i质粒和Cas9/gRNA复合物转入里氏木霉SUS5菌株。理论上除了对eg2基因进行RNAi介导的基因沉默外,还能依赖于CRISPR/Cas9系统对cbh1基因进行敲除。验证引物分别设计在gRNA识别cbh1位点的前后大约250 bp处。从图1可以看出,以出发菌株SUS5作模板扩增出500 bp左右的特异性条带,和预期大小相符;以大部分转化子为模板扩增,虽然扩增出了此特异性条带,但经测序发现和基因组序列完全一致,未能发生有效的基因组编辑。而5号、8号和11号转化子均未能扩增出特异性条带。根据使用Cas9/gRNA复合物对里氏木霉基因组编辑的报道[10],里氏木霉中使用体外组装的Cas9/gRNA编辑基因时主要发生较大DNA片段的插入,这些片段包括染色体DNA、质粒DNA或污染的大肠杆菌的染色体DNA。因此,这些转化子中有可能发生cbh1基因组座位的编辑。
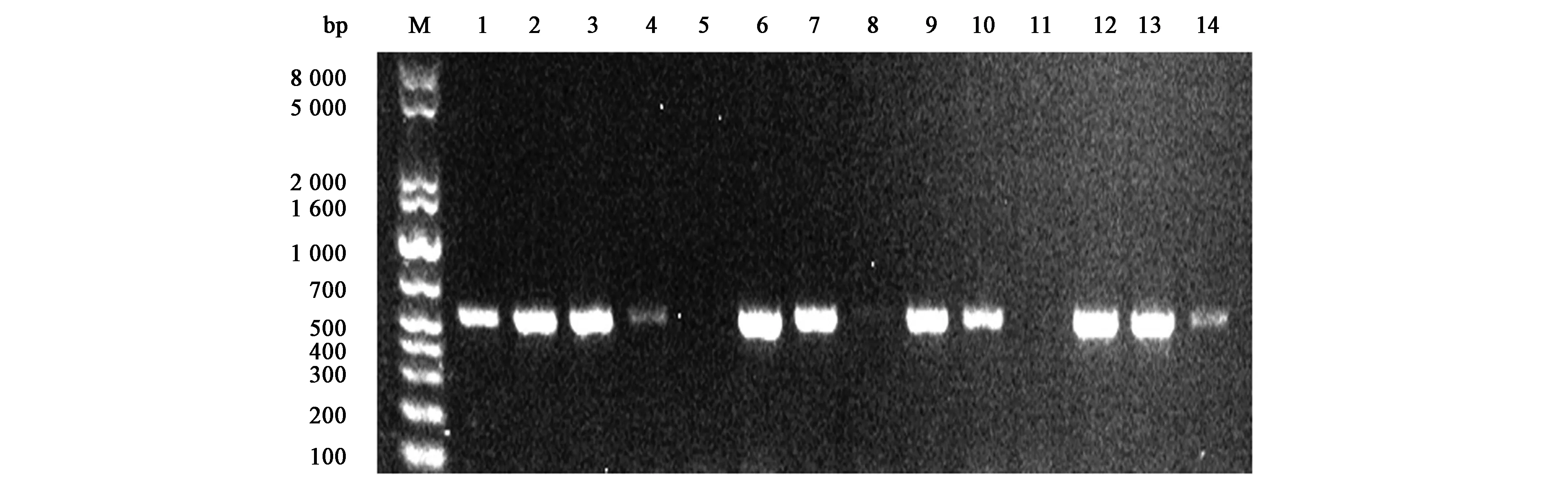
注:M为DNA分子量标准;1~13为转化子;14为SUS5。Note: Lane M is DNA molecular mass marker; lanes 1~13 are transformants; lane 14 is SUS5.图1 转化子中cbh1基因敲除的PCR鉴定Fig.1 PCR verification of cbh1 disruption of T. reesei transformants
2.2 选定转化子中CBH1和EG2分泌表达情况
将5号、8号和11号转化子进行摇瓶发酵,并将转化子发酵第4 d的培养上清(即粗酶液)进行SDS-PAGE分析(图2)。可以看出,11号转化子的粗酶液中没有CBH1蛋白,说明基因组中的cbh1基因已被成功敲除,因此将该菌株命名为SUS6。同时, 11号转化子中的EG2的表达受到了较大程度的抑制。分别测量外切纤维素酶以及内切纤维素酶的酶活,与出发菌株相比,11号菌株的外切纤维素酶以及内切纤维素酶活性均显著降低。因此,相对于出发菌株,该菌主要纤维素酶的表达显著降低甚至完全消失,因此构建的菌株可被认为是低纤维素酶背景的里氏木霉菌株。

注:M为蛋白分子量标准;1为出发菌株SUS5;2~4分别为5号、8号和11号转化子。Note: M is protein molecular mass marker;lane 1 is SUS5;lanes 2~4 are transformant 5, 8, and 11, respectively. 图2 转化子中CBH1和EG2分泌表达情况Fig.2 Secreted expression of CBH1 and EG2 in transformants
2.3 eg2基因转录水平分析
使用荧光定量RT-qPCR方法对eg2基因的表达进行分析,转入Cas9/gRNA蛋白以及RNAi表达质粒的SUS6转化子和出发菌株SUS5的eg2基因的相对转录丰度进行比较。与原始菌株SUS5相比,转化子eg2基因的相对转录丰度下降了约98%,这表明eg2基因的表达受到了显著的抑制;对应于EG2蛋白大大下降的表达量。
2.4 低纤维素酶背景菌株中外源基因NfBgl3A表达分析
以所得到的低纤维素酶背景菌株SUS6为宿主菌,将NfBgl3A的表达质粒转化到该菌中,分别选取两株代表性转化子作摇瓶发酵,测定b-葡萄糖苷酶的酶活并SDS-PAGE分析蛋白的分泌表达情况。如图3所示,以SUS5为宿主菌的两个转化子(SUS5/S7和SUS5/S24),其最高酶活出现在第1 d,分别为11.6和31.9 U·mL-1,随后则迅速随之下降至无法观测;而以SUS6菌株为宿主菌的两个代表性转化子(SUS6/S2和SUS6/S9),其最高酶活分别为172.4和79.3 U·mL-1,分别出现在第2和第1 d。从图3B也可看出,以SUS5为宿主菌表达的NfBgl3A,最高酶活所在的第1 d目的蛋白b-葡萄糖苷酶的条带显著弱于以SUS6菌株为宿主菌的两个代表性转化子。因此,实验说明以低纤维素酶背景的菌株来表达异源基因NfBgl3A,可明显提高目的基因的表达水平。
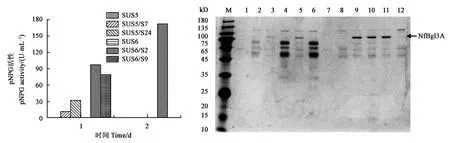
注: M为蛋白分子量标准;1和2为SUS5发酵第1和第2 d样品;3和4为SUS5/S7第1和第2 d样品;5和6为SUS5/S24第1和第2 d样品;7和8为SUS6第1和第2 d样品;9和10为SUS6/S2第1和第2 d样品;11和12为SUS6/S9第1和第2 d样品。Note: Lane M is protein molecular mass marker;lanes 1 & 2 are fermentation supernatants of SUS5 for 1 and 2 d;lanes 3 & 4 are fermentation supernatants of SUS5/S7 for for 1 and 2 d;lanes 5 & 6 are fermentation supernatants of SUS5/S24 for 1 and 2 d;lanes 7 & 8 are fermentation supernatants of SUS6 for 1 and 2 d;lanes 9 & 10 are fermentation supernatants of SUS6/S2 for 1 and 2 d;lanes 11 & 12 are fermentation supernatants of SUS6/S9 for 1 and 2 d.图3 SUS6菌株中NfBgl3A表达增强Fig.3 Enhanced expression of NfBgl3A in the SUS6 T. reesei strain
3 讨论
本研究使用CRISPR/Cas9和RNAi技术相结合,在敲除cbh1基因的同时,还显著降低了宿主菌中主要的内切纤维素酶eg2的表达,从而一步快速得到了低纤维素酶背景的菌株。使用RNAi对目的基因进行表达沉默,目前主要有三种质粒的构建策略:表达反义RNA、使用对向的双启动子和构建表达发卡状RNA的质粒[17]。本研究采用对向双启动子将eg2的一段外显子片段构建到pdc1和eno1强组成型启动子之间,因此质粒转入里氏木霉后,两个启动子会同时对该基因片段进行转录。转录得到的RNA在胞内会相互识别、退火并形成双链RNA结构,为Dicer蛋白剪切为21~25 nt的小RNA片段,指导对目的基因mRNA的剪切。获得的转化子CBH1外切纤维素酶的表达完全消失,而EG2内切纤维素酶的分泌也大大下降,证明在Cas9切割、破坏cbh1基因的同时,RNAi机制还将eg2的mRNA丰度降低了,因此组合CRISPR/Cas9和RNAi技术这种方法能非常有效和快速的获得低纤维素酶背景的里氏木霉工程菌。
使用体外装载Cas9/gRNA对里氏木霉进行基因组编辑,主要发生大片段的染色体或质粒DNA在切割位点的插入。由于有的插入片段可能长度很大, PCR难以扩增出条带,因此,以不能扩增出条带来判定发生了基因组编辑,并进而通过SDS-PAGE来分析表型。据此得到的11号转化子(SUS6)在cbh1基因组座位上发生了基因组编辑,从而不能分泌表达CBH1蛋白。其他两株菌虽然也不能扩增出任何条带,但分析发现,CBH1蛋白仍然被大量表达,因此未能在该基因组座位上发生基因组编辑。
里氏木霉分泌表达大量的内源纤维素酶,这些蛋白需要经过基因的转录、翻译、糖基化、转运等过程才能顺利分泌到胞外。相似的,在里氏木霉中表达外源基因也会同样经历这些过程,因此,如果外源酶和内源纤维素酶受到同样的调控机制,那么,它们在分泌表达时很可能存在一定的竞争。例如,在丝状真菌中进行外源基因的表达常需要使用多拷贝策略,而当启动子拷贝数过多时,则可能出现转录因子对于启动子的“滴定效应”[18],反而降低目的基因的表达。对NfBgl3A的表达发现,在低遗传背景的里氏木霉菌株中表达该酶,不管是酶活还是SDS-PAGE检测,均优于在未破坏纤维素酶表达的出发菌株中进行的实验。NfBgl3A、内源酶cbh1和额外拷贝的eg2基因的表达均使用的是cbh1启动子,因此,NfBgl3A和内源纤维素酶的表达不太可能是在转录调控过程发生了竞争,而更可能是在其他调控途径(例如分泌途径)上有所竞争。但弄清楚相关的分子机制还需要通过更多的实验来加以证明。类似的,在里氏木霉中使用RNAi单独干扰CBH1的表达能提高黑曲霉脂肪酶的表达[19]。值得注意的是,通过对已发表的里氏木霉在纤维素和葡萄糖培养基上的转录组数据分析可以发现,许多已发现的分泌途径关键蛋白所对应基因的转录丰度在纤维素酶表达时并未显著提高[20-21]。因此,如果这些关键蛋白是分泌的限制性因素,当外源基因大量表达时,这些酶就可能和内源纤维素酶竞争分泌通道;而将纤维素酶敲除后,则可提高相应外源基因的表达。
猜你喜欢
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
天津农业科学(2022年6期)2022-07-17
热带农业科学(2022年4期)2022-05-18
干旱地区农业研究(2021年4期)2021-08-11
湖南农业大学学报(自然科学版)(2021年3期)2021-07-02
江西农业学报(2021年4期)2021-04-20
三农资讯半月报(2020年11期)2020-06-21
西夏学(2020年2期)2020-01-24
