
开管离子色谱柱的制备与表征
2020-03-18 06:56黄维雄
色谱 2020年4期
黄维雄
(中国地质大学(武汉)环境学院, 湖北 武汉 430074)
IC一般以低交换容量的离子交换树脂为固定相[5,8],其柱容量通常控制在10-6~10-4eq/mL之间[9](eq表示当量,是早期文献中常用的单位,本文沿用该单位)。与高效液相色谱(HPLC)中广泛使用的硅胶固定相不同,抑制型离子色谱由于通常使用强酸或强碱淋洗液而要求能够耐受pH 0~14的固定相,因此现代IC一般采用有机聚合物固定相以满足耐受极端pH的要求。IC固定相通常由基质和功能基团两部分组成[6]。根据表面功能基与固定相基质的连接方式,可将IC固定相分为接枝型[10]与静电乳胶附聚(electrostatic latex agglomeration, ELA[11])型。接枝型固定相中功能基与基质通过化学键合的方式连接,稳定性较高;而ELA型固定相则通过静电引力作用将带有功能基团的乳胶微球附聚到带相反电荷的基质上,获得非常高的比表面积和交换容量。
开管毛细管液相色谱柱具有体积小、柱压低、柱效高、固定相用量小、试剂消耗少等优点。为保证高柱效,开管液相色谱柱均为毛细管柱,其内径一般小于100 μm[12]。为简便起见,本文从此处起以“开管液相色谱柱”表示开管毛细管液相色谱柱,以“开管离子色谱(open tubular ion chromatography, OTIC)柱”表示开管毛细管离子色谱柱。1979年,Knox等[13]研究了微柱色谱理论并详细讨论了液相色谱毛细管填充柱和开管液相色谱柱的分离效率[13,14],通过理论计算得出开管液相色谱柱的优化内径为0.26 μm,分析时间仅为毛细管填充柱的1/100。如此小内径的开管液相色谱柱在实际应用中难以实现。
OTIC的理论源自开管液相色谱。而作为一种离子分析的专用方法,IC又具有独特性,如固定相为离子交换固定相、淋洗液为酸碱等电解质水溶液,检测器为电导检测器,使用电化学抑制技术降低淋洗液背景等。如果OTIC能够继承常规IC的这些特点,则OTIC将成为可媲美甚至超越常规IC的高效离子分析方法。事实上,随着OTIC柱[15-20]制备技术的发展,非接触式电导检测器(capacitively coupled contactless conductivity detector, C4D)[21-24]和溶液接触式毛细管电导检测器[25]的出现以及毛细管抑制器[18,26,27]的成功研制,抑制型开管离子色谱法已在实验室中成功实现[27]。
由于OTIC研究进展缓慢,相关报道很少,因此目前关于OTIC的综述性报道均包含于毛细管离子色谱[12,28,29]和毛细管液相色谱[30]中。近年来,美国的Dasgupta组在OTIC的研究(特别是分离柱方面)已取得突破性进展[9,20,25,27,31]。而目前国内涉及开管柱的研究多集中在开管液相色谱[32,33]和开管毛细管电色谱[34,35]领域,极少涉及OTIC[29,36]。本文将依据OTIC的发展历程详尽地阐述开管离子色谱柱的制备和表征方法。对OTIC系统中微量进样装置[17,37,38]和检测器[23,24,39-42]等感兴趣的读者可自行查阅相关文献。
1 OTIC柱的制备
本文依据OTIC柱制备的发展历程,将OTIC柱按基质材料分为二氧化硅基质和有机聚合物基质两大类。
1.1 二氧化硅基质的OTIC柱
1981年,Ishii等[15]制备了磺酸型阳离子交换OTIC柱:30~60 μm i.d.的玻璃毛细管内表面先经苯基三乙氧基硅烷或者2-巯基乙基三乙氧基硅烷化学修饰,再通过浓硫酸或者高锰酸盐氧化以获得离子交换基团。所得的开管阳离子分离柱的柱容量为2×10-8eq/m。实验表明增加内表面积可有效增大柱容量,升高柱温可明显地提高柱效。1987年,Pfeffer等[43]将氯化十六烷基吡啶动态修饰到C8反相开管柱上,制成阴离子交换开管柱。
1983年,Manz等[44]制备了三氯十八烷基硅烷修饰的二氧化硅开管柱(25 μm i.d.)。1989年,Muller等[45]报道了更小内径(9.5、5.4和4.6 μm)的聚丁二烯磺酸或聚丁二烯马来酸修饰的阳离子交换二氧化硅开管柱,成功分离了碱金属、碱土金属离子、6种氨基酸和4种核苷。为了制备厚度约0.1 μm的固定相,作者采用静态真空加温干燥技术制备开管柱,真空加温干燥时间长达6个星期。1991年该课题组又采用静态与动态相结合的开管柱制备技术[16],分别制备了固定相厚度更薄的阳离子交换开管柱和阴离子交换开管柱:4.6 μm i.d.的阳离子交换开管柱可在25 s内分离碱金属离子;相同内径的阴离子交换开管柱在35 s内即可分离6种常见的一价阴离子;2.3 μm i.d.的二氧化硅毛细管内表面不需要任何化学修饰,仅需控制pH 9.4,利用毛细管内表面自带的硅醇基即可在70 s内实现Li+/Na+(共淋洗)、K+、Rb+和Cs+等阳离子的快速分离。但是随着柱内径的减小,开管柱的制备难度显著增加,如越来越容易发生堵塞等[19];另外,使用硅烷化学修饰的阳离子交换和阴离子交换二氧化硅开管柱在高pH条件下不稳定[16,18,19]。
在液相色谱中,溶质在固定相和流动相中的扩散系数相当,增加固定相厚度所导致的柱效损失较气相色谱而言要小得多[13]。为了增加柱容量以及减少开管柱制备时间,可使用75 μm i.d.的二氧化硅毛细管制备多层固定相修饰的开管柱[18,26,46]。在制备二氧化硅开管柱之前,通常需要对二氧化硅毛细管内表面进行化学预处理[16,17],例如先用HF/HNO3(2.5%, v/v)腐蚀毛细管内壁30 min,再用HCl(1%, v/v)清洗30 min,最后用去离子水清洗至pH中性[17]。目的是获取尽可能多的硅醇基以增强固定相的附着力。
2007年,Kuban等[18]利用双环氧化合物和伯胺的缩聚反应,采用动态缩聚技术,制备了阴离子交换固定相修饰的开管柱(75 μm i.d.)。制备过程如下:(A)将2.8%甲胺和7.2% 1,4-丁二醇二缩水甘油醚(1,4-butanedioldiglycidyl ether, BDDE)的混合水溶液持续泵入预处理后的二氧化硅毛细管30 min,获得黏附在毛细管内壁的“第一层”; (B)依次泵入4%甲胺水溶液30 min, 10% BDDE水溶液30 min; (C)重复n-1次步骤(B)。每层固定相的制备时间约1 h。固定相的结构见图1。扫描电镜实验测得7层固定相的厚度约200 nm。采用逐层动态缩聚方法的优点是可通过逐层性能表征以确定合适的缩聚层数。作者通过该法制备了多达25层的多孔型固定相。实验表明柱容量随着缩聚层数的增加而增加。将该开管柱与管状抑制器连接,以1.0 mmol/L KOH为淋洗液,采用C4D检测,首次获得了抑制型开管离子色谱图。但是由于该开管柱的内径为75 μm,需要长达5 m的柱长才能得到满意的柱效,且所获得的离子色谱图的死时间太长。2011年,Huang等[26]采用类似的缩聚技术,制备了多层壳聚糖修饰的阴离子交换开管柱(75 μm i.d.)。
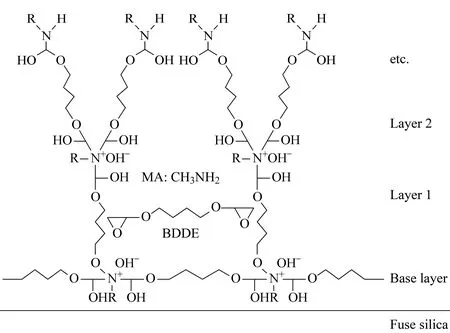
图1 聚合物结构[18]
2008年,Kuban等[46]制备了以多层聚丁二烯-马来酸(poly(butadiene-maleic acid), PBMA)为固定相的阳离子交换开管柱。单层固定相制备过程如下:先将PBMA聚合物前体与5%的偶氮二异丁腈的混合物泵入预处理过的二氧化硅毛细管,再用N2将柱中的混合液吹出,仅在毛细管内壁留下一层很薄的液膜;然后将含液膜的毛细管置于烘箱中进行热聚合反应(160 ℃, 15 min)。多层固定相可通过同样的方式逐层叠加制备。实验发现单层固定相的厚度取决于PBMA的使用浓度。两层PBMA修饰的二氧化硅开管柱(1 m×75 μm i.d.)可通过重力流以1 mmol/L酒石酸为淋洗液在10 min内分离4种碱金属阳离子。
一种更为简便的开管柱制备方式是采用ELA技术,将乳胶微球固定相静电吸附到带相反电荷的基质上。该技术具有快速传质的优点[19],可追溯到最早的抑制型离子色谱报道[1]。随着乳胶微球制备技术的发展,ELA已成为一种IC固定相制备的常用技术[19,47,48]。由于酸处理后的二氧化硅毛细管内表面的硅醇基在一定的pH条件下会电离而使其内表面带负电,因此带正电的乳胶微球极易静电吸附到二氧化硅毛细管的内表面,形成单层阴离子交换固定相。同种乳胶微球由于带同种电荷而互相排斥,因此只能单层吸附。
1997年,Pyo等[17]将粒径为375 nm的阴离子交换乳胶微球静电吸附到二氧化硅毛细管(3 m×50 μm i.d.)内表面,制成阴离子交换开管柱。制备过程非常简单:直接将稀乳胶微球悬浊液持续泵入预处理过的二氧化硅毛细管,再用去离子水冲洗毛细管以除去毛细管中未吸附的乳胶微球即制成开管柱。实验发现用该法制得的单层乳胶微球附聚的开管柱已具备较大的柱容量。为进一步增大柱容量,可对二氧化硅开管柱进行阴离子交换乳胶(A)和阳离子交换乳胶(B)微球的交替吸附,如形成A-B-A-B-A 5层吸附结构[18]。实验表明通过这种交替吸附方式所制得的开管柱的柱容量虽然比单层吸附更高,但却损失了柱效。2012年,Diao等[36]也采用该方法制备了多层乳胶交替吸附的二氧化硅开管柱。将ELA技术应用于二氧化硅开管柱的不足在于:高pH的淋洗液会使乳胶微球从毛细管的内表面整体脱落,损坏开管柱。
ELA技术也用于制备开管毛细管电色谱柱[49]、预浓缩柱[50]和硅胶整体柱[51]。2000年,Breadmore等[49]将AS5A(75 nm粒径)乳胶微球静电吸附到二氧化硅毛细管内表面用于无机阴离子的毛细管电色谱分离。2006年,Zhang等[50]将AS5A和CS3(300 nm粒径)乳胶微球依次吸附到二氧化硅毛细管内表面,制成双层离子交换乳胶吸附开管柱(见图2),用于阳离子的毛细管电色谱预浓缩和电泳分离。
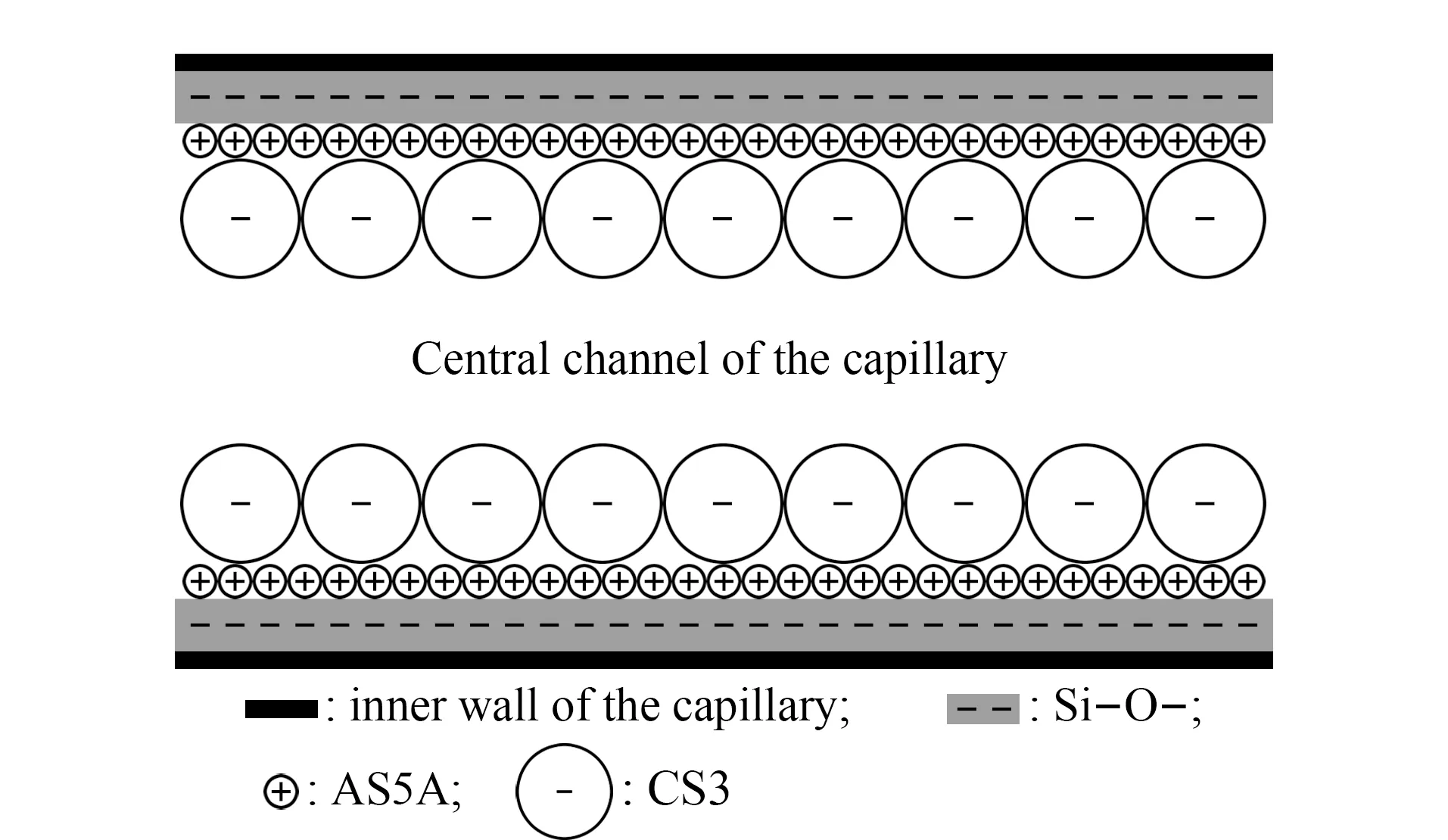
图2 双层乳胶附聚二氧化硅开管柱结构示意图[50]
1.2 有机聚合物基质的OTIC柱
采用ELA技术制备开管柱的最大优点是易制备和耗时短(可低于30 min)。除了二氧化硅以外,很多有机聚合物的表面也容易经氧化/水解/磺化而使其表面带负电[19],因此也可以将带正电的乳胶微球吸附到经化学预处理的有机材质毛细管内壁上。
在经典的采用抑制电导检测的阴离子交换色谱法中,当淋洗液为OH-时,经抑制后的淋洗液是极弱电离的纯水,这极大地降低了淋洗液背景电导,从而降低了噪声水平,提高了信噪比,降低了检出限;同时通过抑制器引入极限摩尔电导值更高的对离子,进一步提高了被分析离子电导检测的灵敏度。因此抑制电导检测比直接(非抑制)电导检测灵敏度更高,检出限更低。
鉴于抑制电导检测灵敏度高、检出限低的优点以及ELA型开管柱容易制备的特点,Dasgupta组一直致力于研制兼容OH-淋洗液的ELA型阴离子交换开管柱,并结合抑制电导检测,实现抑制型开管离子色谱。
首先是兼容OH-淋洗液的ELA型开管柱的制备。早期的尝试包括ELA型二氧化硅开管柱(50 μm i.d.)的制备[17]。经测试发现该开管柱不兼容OH-淋洗液[18]:低浓度的OH-淋洗液(<5 mmol/L)在几分钟内就会使开管柱中的乳胶微球脱落,损坏分离柱。不兼容的原因可能如下:硅醇基的酸性极弱,吸附乳胶微球时未完全电离,导致其与乳胶微球间静电引力太弱;淋洗液中的氢氧根腐蚀二氧化硅基质。
为此,Dasgupta组[19,20]尝试使用有机聚合物材质的毛细管:Zhang等[19]采用ELA技术制备了聚甲基丙烯酸甲酯(polymethylmethacrylate, PMMA)基质的阴离子交换开管柱(16~20 μm i.d.),使用3种无损方法测定该开管柱的柱容量,并搭建了一套非抑制型开管离子色谱系统[37]。PMMA开管柱的制备过程如下:(A)毛细管内表面预处理:取一段约80 cm的PMMA毛细管,先用乙醇冲洗30 min,清除柱内可能残留的有机物,再用去离子水冲洗毛细管,然后持续泵入NaOH溶液(10%, w/v)90 min(利用强碱水解毛细管内表面的酯基以获得带负电的-COO-),最后再用去离子水冲洗毛细管至pH中性。(B)乳胶附聚:用两个气动泵将稀释10倍的乳胶悬浊液正反向交替(每个方向10 min)泵入PMMA毛细管至少2 h,然后用一个气动泵单方向泵入去离子水,置换出未吸附的乳胶悬浊液。实验表明使用1 mmol/L苯甲酸钠淋洗液,该开管柱对Cl-的柱效可达到1.5×105/m;但是当使用≥ 10 mmol/L OH-淋洗液一段时间后,乳胶微球仍然从PMMA开管柱的内表面脱落[19,20]。原因可能是淋洗液中的OH-腐蚀PMMA基质(PMMA在碱性条件下易水解)。
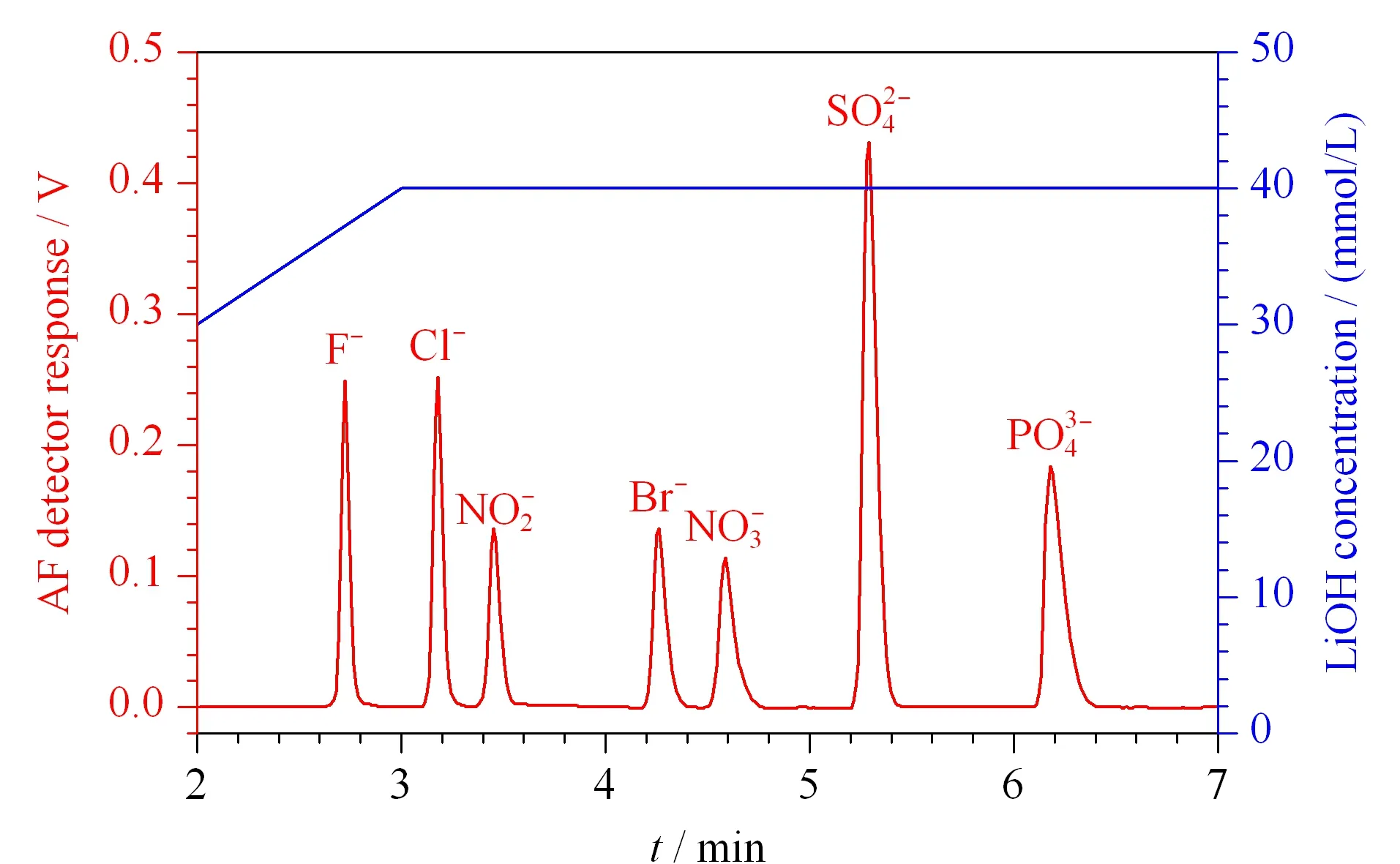
图3 AS18乳胶附聚COP开管柱梯度色谱图[27]
Huang等[20]进一步尝试了环烯烃聚合物/共聚物(cycloolefin polymer/copolymer, COP/COC)材质的毛细管,成功制备了兼容OH-淋洗液的ELA型阴离子交换COP开管柱(19~28 μm i.d.)。制备过程如下:(A)毛细管内表面氯磺酸(-SO3Cl)化:先用干燥的N2吹洗毛细管>5 min以清除毛细管中的水分,然后用气动泵将85%~95%/15%~5%(质量分数)氯磺酸/冰醋酸混合液泵入干燥的COP毛细管2~15 h,磺化毛细管内表面。由于磺化过程中氯磺酸过量,因此COP内表面的-SO3H会进一步与氯磺酸反应,生成-SO3Cl[52]。(B)-SO3Cl水解成-SO3H:先用气动泵持续将冰醋酸泵入COP毛细管30 min以置换出毛细管中未反应的ClSO3H,然后用去离子水在较高的恒定流速下(1.5~10 μL/min)冲洗毛细管4~5天,使毛细管内表面的-SO3Cl完全水解成-SO3H。(C)乳胶附聚:按PMMA开管柱的乳胶附聚步骤完成COP开管柱的制备。实验发现该COP开管柱兼容40 mmol/L的OH-淋洗液,因此适合OH-等度或者梯度淋洗。此外,Huang等[27]还成功研制了配套的电渗析毛细管抑制器,与COP开管离子色谱柱配合使用,得到了兼容OH-淋洗液的抑制型开管阴离子色谱(图3为7种常见阴离子的梯度色谱图)。虽然COP开管柱的制备周期较长(>5天),但它是迄今为止已报道的唯一兼容OH-淋洗液的乳胶附聚型开管柱。
2 OTIC柱的表征
2.1 柱容量的实验测定
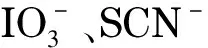
自2013年起,Dasgupta组利用ELA技术先后制备了10~30 μm i.d.的PMMA[19]和COP[20]离子色谱开管柱,并测定了乳胶微球附聚前后的阳离子交换和阴离子交换容量[19,31]。由于PMMA和COP开管柱的柱容量低,测量准确度差,因此在柱容量测定时使用了3种不同的测量方法并相互比对测量结果。这3种柱容量测量方法分别为:(A)荧光素离子置换法(fluorescent ion displacement, FID); (B)迎头置换色谱法(frontal displacement chromatography, FDC); (C)迎头反应色谱法(frontal reaction chromatography, FRC),也称柱上酸碱滴定法。其中FID法为离线测定法,较耗时;而FDC和FRC法均为柱上测定方法,简便可靠。通过选择合适的探针离子,FDC和FRC法可使用光度或C4D检测指示滴定终点阶跃突变。对AS18乳胶附聚的PMMA开管柱,实验发现FRC法测得的柱容量约为FID和FDC测得值的2倍,原因为AS18乳胶微球含有大致等量的强碱性和弱碱性功能基团,FRC法测得功能基团总量,而FID和FDC仅测定强碱性功能基团[19]。对FRC法,可使用以下公式计算开管柱的柱容量[31]:
CAEX=CH+×Q×(ts-th)=CH+×Q×Δt
(1)
CCEX=COH-×Q×(ts-th)=COH-×Q×Δt
(2)
其中CAEX和CCEX分别为阴离子交换开管柱和阳离子交换开管柱的柱容量,CH+和COH-分别为酸、碱滴定剂的浓度,Q为滴定剂的体积流速,ts和th分别为滴定终点的阶跃突变时间和开管柱的死时间,Δt为ts和th之间的时间差。FRC法测定CAEX步骤如下:先用气动泵将强碱溶液泵入阴离子交换开管柱30 min,把乳胶微球中的可交换阴离子全部转换成OH-,再用去离子水冲洗开管柱30 min,最后用气动泵将1 mmol/L强酸滴定液在恒定的低流速下泵入开管柱,用C4D或UV柱上检测(数据记录自柱上滴定起始至滴定终点阶跃突变出现后为止),同时以重量法测定流速。
对PMMA开管柱[19],用强碱进行水解处理后所测得的CCEX为0.6~1.5 peq/mm2(为方便不同内径开管柱之间的柱容量比较,以开管柱中单位内表面积的离子交换当量表示开管柱的柱容量);经AS18乳胶附聚后的CAEX为12~20 peq/mm2。而用氯磺酸磺化后的COP开管柱[20]的CCEX可高达300 peq/mm2;经AS18乳胶附聚后的CAEX则为20~30 peq/mm2,较PMMA开管柱大,其原因可能是氯磺酸腐蚀了COP毛细管的内表面,增大了内表面积,但仍按腐蚀前的内表面积计算柱容量。
测定柱容量时如果信号突变太平缓(如信号变化的时间窗口太宽或柱容量太小,后者在测定COP磺化后的阳离子交换容量时经常遇到),将导致ts值难以界定,如图4b中的红实线所示。通过对该类数据求取一阶导数,则通常可得到类似的色谱峰形(图4b中的红虚线)[31],此时即可根据峰值出现的时间确定ts值,从而降低测量误差。
2.2 柱容量的理论计算
对单层乳胶附聚型的离子交换填充柱和开管柱,其柱容量均可通过乳胶微球的粒径和单个乳胶微球的离子交换容量进行估算,并用来与实测值进行比较[9]。阴离子交换乳胶微球的比容量s在几十到几百μeq/g之间,密度ρ=1.1~1.2 g/cm3,粒径dlp=50~200 nm。单个乳胶微球的离子交换容量Clp计算式如下:
(3)
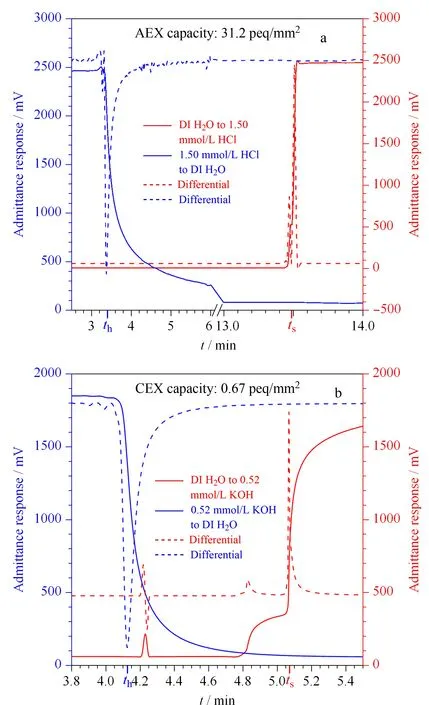
图4 乳胶附聚前后的COP开管柱的(a)阴离子交换和(b)阳离子交换容量测定(虚线代表一阶求导数据)[31]
其中Clp以zeq为单位(zeq: 10-21eq)。Clp乘以阿伏伽德罗常数即可得单颗乳胶微球的离子交换位点数(nlp;以一价计):
(4)
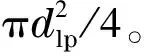
(5)
然而,由于分子孔隙度测定法提供的表面积测定结果不能等同于可静电吸附乳胶微球的表面积,因此填充柱的柱容量应以实测值为准[54]。乳胶微球的投影面积与微球直径的二次方成正比,而方程(3)显示单颗乳胶微球的柱容量与微球直径的3次方成正比,因此将方程(3)代入方程(5)可得柱容量Cpc,max与乳胶粒径dlp成正比,即可通过控制乳胶微球的粒径来调节柱容量。
对长为L,内径为dc的开管柱,其内表面积Stot=πdcL,将其代入方程(5)即可得开管柱的柱容量最小值COT,min(称其为最小值是因为开管柱预处理时内表面发生了化学腐蚀,因而增大了有效内表面积,但仍按腐蚀前的内表面积计算柱容量):
(6)
对AS18乳胶(Clp=34 zeq,dlp=65 nm)附聚的COP开管柱(dc=19 μm,L=951 mm),计算得到其柱容量COT, min=0.58 neq/柱。而经FRC实验测得的COT, min值为(2.31±0.12) neq/柱(n=3),是计算值的4倍[54]。因此,柱容量应以实测值为准。
2.3 固定相相同时填充柱与开管柱的联系
ELA技术同时被用于填充柱[47,55,56]和开管柱[20]的制备。当使用同种乳胶微球固定相时,虽然填充柱和开管柱的柱容量相差可达5个数量级以上,但是两者之间必然存在一定的联系。Huang等[9]定义了新的柱容量表示参数,经理论推导并通过实验验证了如下关系:当使用同种固定相时,由填充柱的色谱保留行为可以预测开管柱的色谱行为;反之亦然。
填充柱的柱容量一般以μeq/柱或μeq/mL为单位[3]。为了方便比较,Dasgupta在研究开管柱的时候除了用neq/柱表示柱容量以外,为找出一个与开管柱内径无关的参数,还将柱容量表示成αiex:单位表面积上的离子交换当量,单位peq/mm2[19]。为了寻找填充柱与开管柱之间的联系,使用了离子交换相比βiex:相同柱长条件下淋洗液中的离子当量(eq/cm)与固定相的离子交换容量(eq/cm)的比值;但在计算βiex时,须预先指定淋洗液浓度[19,20]。为找出一个与淋洗液浓度无关的参数,Dasgupta从淋洗液的当量浓度是分离柱中淋洗液体积的函数出发,提出了另一个表示柱容量的参数γiex:离子交换分离柱中单位体积淋洗液所对应的固定相离子交换当量。对开管柱,γiex和αiex存在着简单的关系[9]:
(7)
对同种乳胶附聚的填充柱(柱1)和开管柱(柱2),等度淋洗条件下柱1和柱2中被测离子的保留因子均与各分离柱自身的柱容量成正比,此时应有如下关系式:
(8)
其中γiex1和γiex2分别为柱1和柱2的柱容量;ki,1和ki,2分别为柱1和柱2中被测离子i的保留因子。
而离子色谱中被测离子的保留因子k与淋洗液浓度[E]之间存在如下关系:
log10k=-alog10[E]+b
(9)
其中a为被测离子所带的电荷数,b为常数。
对同种被测离子,k/γiex值应与柱型无关,因此使用同种乳胶固定相的填充柱和开管柱应具有相同的log10k/γiex-log10[E]直线,如图5所示[9],对同种离子,图中显示的两根直线存在一定的斜率差和截距差,其原因包括:填充柱和开管柱的实验数据来自不同的实验室,且在不同的温度下测得;开管柱中KOH淋洗液为手动配制,难以避免CO2的影响等。

图5 相同固定相的填充柱和开管柱的log10k/γiex-log10[E]图[9]
2.4 柱效
2.4.1开管柱的van Deemter曲线绘制
液相色谱开管柱的理论塔板高度H可通过下式计算[37]:
(10)
其中L为柱长,w1/2、tR分别为被分离组分的半峰宽和保留时间。开管柱的van Deemter方程表达式如下[19,20]:
H=2Dm/u+Cu
(11)
其中u为淋洗液的线流速,Dm为溶质的扩散系数,C为常数项。Dm和C可通过对实验数据拟合得到:先通过实验获得不同u时某一离子(如Cl-)的色谱图,再根据方程(10)计算出相应的H值,绘制H-u散点图,然后对数据进行拟合,得到Dm和C的最佳值[57,58]。这两个参数的标准偏差可通过“jackknife”方法求得[59]。令dH/du=0,可进一步求得拟合曲线最低点的umin和Hmin值(见图6)[37]。
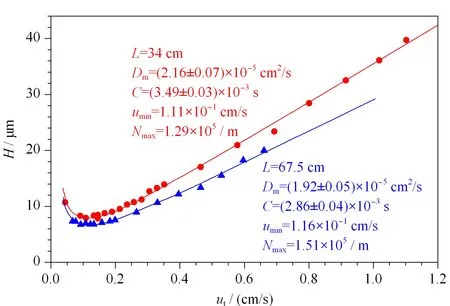
图6 乳胶附聚PMMA开管柱的van Deemter曲线[37]
2.4.2高温OTIC
有两种方法可有效提高液相色谱开管柱的柱效:一是减小开管柱的内径;二是升高柱温[17,60]。减小柱内径意味着更小的进样体积和检测体积(当检测器为柱上检测器时),不容易实现。而升高柱温则相对简便的多。在一个液相色谱开管柱系统中,为了减少分析时间,将柱温从20 ℃升到200 ℃所产生的效果等同于将柱内径从50 μm降到12 μm(502/122=17.36),原因在于溶质的扩散系数随着温度的升高而升高[60],其理论依据来自Stokes-Einstein方程[61]:
(12)
其中kB为Boltzmann常数,T为绝对温度,a为溶液中溶质的半径,η为淋洗液的黏度。水相中溶质在200 ℃时的扩散系数Dm200与其在20 ℃时的扩散系数Dm20的关系[60]为Dm200/Dm20=(η20/η200)×(473/293)=17.47≈17.36。
Pyo等[17,61,62]制备了乳胶附聚的二氧化硅OTIC柱(50 μm i.d.),通过实验证实升高温度虽然缩短了被分析离子的保留时间,但是显著地提高了柱效,使van Deemter曲线更平缓。升高温度除了增大离子的Dm值、提高柱效以外,还能改变离子的选择性[63]。需要注意的是:对固定相中含季铵功能基的阴离子交换开管柱,使用时温度不宜超过100 ℃,因为在碱性介质条件下,高温会使季铵功能基团快速发生去烷基化的Hoffmann消除反应,损失柱容量[12,46]。
2.5 开管柱的均一性表征
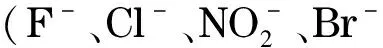
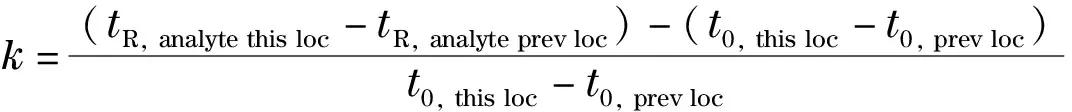
(13)
其中tR, analyte this loc和tR, analyte prev loc分别为被测离子在当前位置和先前位置的保留时间,t0, analyte this loc和t0, analyte prev loc则分别为当前位置和先前位置的死时间。此外,可通过相同淋洗液条件下C4D检测池在COP柱上不同位置的背景响应来获取COP柱内径的均匀度信息。实验表明乳胶微球固定相在COP开管柱中分布较均匀,且不受磺化度(当磺化度低时)的影响。与自动化的全柱检测技术[64]相比,以上提到的3种开管柱均一性表征实验方法更加简便易行。
3 结论与展望
IC分离柱必须耐强酸、强碱的特性促进了有机聚合物基质OTIC柱的研制。ELA技术极大地方便了离子色谱开管柱的制备。兼容OH-淋洗液的阴离子交换COP开管柱的成功研制使抑制型开管离子色谱的梯度洗脱得以实现。目前OTIC需要解决的主要问题是改善重现性,进一步的研究包括尝试使用其他有机聚合物基质的毛细管制备开管柱,毛细管抑制器的改进和nL/min级的淋洗液生成器的研制等。利用开管柱质量轻、耐受温度范围宽等特点,未来OTIC可制成微型便携式仪器应用于复杂环境的现场监测和地外星球探索等,应用前景广阔。
猜你喜欢
保定学院学报(2022年6期)2022-12-01
今日农业(2022年14期)2022-09-15
保健与生活(2022年11期)2022-06-09
老年博览·上半月(2021年6期)2021-07-01
陶瓷学报(2020年5期)2020-11-09
天津医科大学学报(2019年3期)2019-08-13
中成药(2018年2期)2018-05-09
中国粮油学报(2016年1期)2016-02-06
杂草学报(2015年2期)2016-01-04
中药与临床(2015年5期)2015-12-17
