
三氟啶磺隆的合成
2020-03-19 06:57丁华平陈素云
世界农药 2020年2期
丁华平,陈素云
(江苏长青农化股份有限公司,扬州 225200)
三氟啶磺隆是先正达公司于2001年开发的高效磺酰脲类除草剂,化学名称为1-(4,6-二甲氧嘧啶-2-基)-[3-(2,2,2-三氟乙氧基)-2-吡啶磺酰基]脲,主要通过抑制植物体内的乙酰乳酸合成酶的活性,影响支链氨基酸的合成,从而抑制杂草生长达到灭草效果。
三氟啶磺隆可以由中间体3-(2,2,2-三氟乙氧基)吡啶-2-磺酰胺和2-氨基-4,6-二甲基嘧啶偶联得到。后者可以大量得到,前者目前有3条合成路线。合成路线一是选用2-氯-3-羟基吡啶为起始原料,首先选用苄基保护羟基,然后和硫脲反应,接着通入氯气氧化再氨化,最后在钯碳的催化下氢解脱保护得到3-三氟乙氧基吡啶-2-基磺酰胺。合成路线二同样是选用2-氯-3-羟基吡啶为起始原料,首先与对甲苯磺酸三氟乙酯反应,再与硫脲反应得到2-巯基-3-三氟乙氧基吡啶,通入氯气后氨解得到3-三氟乙氧基吡啶-2-基磺酰胺。合成路线三是以烟酰胺为起始原料,首先发生Hofmann降解得到3-氨基吡啶,然后被过氧化氢氧化后氯代得到2-氯-3-氨基吡啶,重氮化后再用三氟乙醇醇解得到2-氯-3-三氟乙氧基吡啶,再与硫氢化钠反应得到2-巯基-3-三氟乙氧基吡啶,用次氯酸钠氧化得到3-三氟乙氧基吡啶-2-基磺酰胺。3条路线相比,合成路线一选用苄基保护羟基,而且脱保护要用贵金属催化,后两条路线步骤较短,不用保护羟基,直接反应。
在已有的合成路线的基础上,笔者同样选用不保护羟基的方法,以2-硝基-3-羟基吡啶为起始原料,首先和甲磺酰氯反应,然后和苄硫醇发生亲核取代,和2,2,2-三氟乙基甲磺酸酯发生醚交换,再经过次氯酸钠的氧化,氨气氨化得到目标中间体,最后和2-氨基-4,6-二甲基嘧啶偶联得到三氟啶磺隆。

图1 三氟啶磺隆的合成路线
1 试验部分
1.1 主要仪器与试剂
BrukerAVANCE(核磁共振波谱仪600 MHz)和Agilent 400 MHz;maXis超高分辨飞行时间质谱仪(maXis)。
所用试剂无特殊说明均为市售分析纯试剂,由国药试剂或萨恩化学技术(上海)有限公司提供。
1.2 三氟啶磺隆的合成
1.2.1 2-硝基吡啶-3-基甲磺酸盐的合成
在50 mL的茄形瓶中加入2-硝基-3-羟基吡啶(1.4g,10mmol)、三乙胺(1.39mL,1.3eq)和10mL二氯甲烷,将所得溶液冷却至0℃,然后缓慢滴加甲磺酰氯(1 mL,1.3 eq)。滴加完毕后,温度升高至室温反应1.5 h,反应结束后加入少量稀HCl和NaHCO3溶液洗涤,用乙酸乙酯萃取,得到的有机相用无水硫酸钠干燥后旋干,得到化合物1为深黄色油状物,质量为2.02 g,不作进一步提纯。
1.2.2 2-(苄硫基)吡啶-3-基甲磺酸盐的合成
在50 mL的茄形瓶中加入化合物1(2.02 g)、苄硫醇(1.29 mL,1.1 eq)和15 mLN,N-二甲基甲酰胺,冰浴下搅拌。将氢氧化钾(1.9g,1.4eq)溶解于4.4mL H2O中制备30%的氢氧化钾水溶液,冰浴下将氢氧化钾溶液缓慢滴加至上述溶液中。滴加完毕后继续冰浴反应1 h,反应结束后将混合液倒入盐水中,用乙酸乙酯萃取,得到的有机相用无水硫酸钠干燥后旋干,柱层析得到化合物2为淡黄色固体,质量为1.51 g,前两步总产率为51%。试验数据:1H NMR(400 MHz,CDCl3)δ 8.41(dd,J=4.7,1.4 Hz,1H),7.58(dd,J=8.1,1.4 Hz,1H),7.38(d,J=7.2 Hz,2H),7.32-7.26(m,2H),7.25-7.23(m,1H),7.08(dd,J=8.1,4.8 Hz,1H),4.47(s,2H),3.17(s,3H).
1.2.3 2-(苄硫基)-3-(2,2,2-三氟乙氧基)吡啶的合成
在50 mL的茄形瓶中加入化合物2(1.48 g,5 mmol)、2,2,2-三氟乙基甲磺酸酯(1.6 g,1.8 eq)、碳酸钾(1.03 g、1.5 eq)、10 mL N,N-二甲基甲酰胺,在120℃油浴下反应2 h。反应结束后,加入水淬灭,用乙酸乙酯萃取,得到的有机相用无水硫酸钠干燥后旋干,柱层析得到化合物3为白色固体,质量为1.2 g,产率为80%。试验数据:1H NMR(400 MHz,CDCl3)δ 8.20(dd,J=4.2,1.9 Hz,1H),7.44-7.40(m,2H),7.33-7.28(m,3H),7.26-7.21(m,2H),7.02-6.99(m,2H),4.44(s,2H),4.35(q,J=8.1 Hz,2H)。
1.2.4 3-(2,2,2-三氟乙氧基)吡啶-2-磺酰胺的合成
在100 mL的茄形瓶中加入化合物3(1.49 g,5 mmol)、15 mL二氯甲烷、10 mL浓盐酸,将所得溶液冷却至-5℃,然后缓慢滴加有效氯含量为8%的次氯酸钠(18.5 mL,10 eq),滴加完毕后室温搅拌1.5 h,反应结束后,用乙酸乙酯萃取,得到的有机层用无水硫酸钠干燥,旋干后加入10 mL二氯甲烷,冷却至0℃,然后通入氨气20 min,旋干溶剂,柱层析得到化合物4为白色固体,质量为0.78 g,产率为61%。试验数据:1H NMR(400 MHz,DMSO)δ 8.26(d,J=4.4 Hz,1H),7.83(d,J=8.1 Hz,1H),7.64(dd,J=8.5,4.5 Hz,1H),7.31(s,2H),4.99-4.91(m,2H).13C NMR(101 MHz,DMSO)δ 151.01,148.63,141.40,128.53,123.95,122.65,65.79。
1.2.5 三氟啶磺隆的合成
在50mL的茄形瓶中加入化合物4(1.28g,5mmol)、4,6-二甲氧基-2-(苯氧基羰基)氨基嘧啶(1.6 g,1.2 eq)、10 mL乙腈,冰浴降温至0℃并缓慢滴加三乙胺(0.8 mL,2 eq),滴加完毕后室温下搅拌1 h,反应结束,旋干溶剂,残液中加入少量稀HCl洗涤,然后用乙酸乙酯萃取,有机层用无水硫酸钠干燥后柱层析得到化合物5,质量为1.88 g,产率为86%。试验数据:1H NMR(400 MHz,DMSO)δ12.75(s,1H),10.60(s,1H),8.31(s,1H),7.88(d,J=8.6 Hz,1H),7.73(d,J=7.5 Hz,1H),5.96(s,1H),4.99(q,J=9.1 Hz,2H),3.85(s,6H).13C NMR(101 MHz,DMSO)δ172.04,158.26,157.79,151.41,150.89,141.70,127.18,125.64,81.43,66.61,54.33。
2 结果与讨论
在该路线的合成步骤中,最为关键的是第4步化合物4的合成。这一步是氧化反应,选用的氧化剂是次氯酸钠,首先苄硫基团和次氯酸钠发生氧化还原反应,生成亚磺酸中间体,然后氯离子进攻取代得到磺酰氯,因此次氯酸钠作为氧化剂需要过量,得到的中间体磺酰氯再被氨气氨化得到化合物5。首先,在盐酸加入量和反应时间不变时,于-5℃下改变次氯酸钠的加入量,考察次氯酸钠的用量对磺酰氯产率的影响,见表1。

表1 次氯酸钠用量对磺酰氯产率的影响
试验发现,随着次氯酸钠用量的增加,反应产率得到了提高,当次氯酸钠的加入量是反应物的10倍时,生成磺酰氯的产率最高,达到74%,继续提高次氯酸钠的用量对反应产率的影响并不大。
由于这一步是氧化反应,在滴加次氯酸钠过程中,会出现反应体系温度升高的情况,一旦温度升高,就会发生副反应。为了防止副反应对磺酰氯产率的影响,笔者采用低温缓慢滴加的方法。在盐酸、次氯酸钠加入量和反应时间不变时,考察了滴加温度对于磺酰氯产率的影响(表2)。
试验发现,当滴加温度高于0℃时,反应产率较低,可能有副反应发生,当温度降低至-5℃以下时,能有效抑制副反应的发生,产率提高至74%。因此,最终选择滴加温度为-5℃。

表2 滴加温度对反应产率的影响
笔者首先尝试将得到的磺酰氯缓慢滴加至氨水,发现反应效果不佳,原料在长时间的反应条件下都不能反应结束,因此选择将得到的磺酰氯溶解在二氯甲烷中,然后通入氨气氨化,氨化的过程很快,效率很高,因此需要控制通入氨气的时间。在恒定温度为0℃时,通入不同时间的氨气,考察氨化时间对磺酰氯产率的影响(表3)。
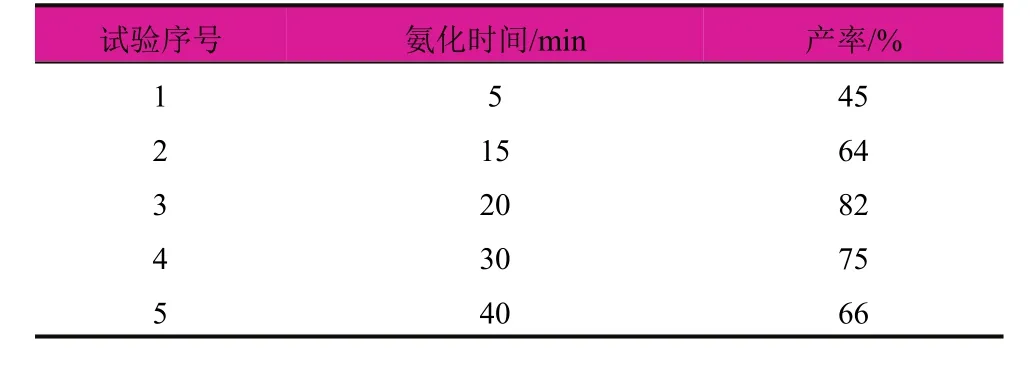
表3 氨化时间对磺酰氯产率的影响
试验发现,氨化时间为20 min时的效果最好,收率能够达到82%,继续延长反应时间,反而会使产物转化为杂质,收率降低。
3 结 论
本合成路线主要选用2-硝基-3-羟基吡啶为起始原料,首先经过两步取代反应,然后发生醚交换,再氧化、氨化,最终偶联得到产物三氟碇磺隆。本合成路线反应条件易于达到,可工业化应用。
猜你喜欢
现代农业科技(2022年19期)2022-11-21
工业催化(2022年9期)2022-10-17
河南农业科学(2022年6期)2022-09-03
科学导报(2022年21期)2022-04-10
食品安全导刊(2021年20期)2021-08-30
西北农林科技大学学报(自然科学版)(2021年1期)2021-03-04
农药科学与管理(2020年2期)2020-04-22
北京大学学报(自然科学版)(2019年6期)2019-11-27
成长·读写月刊(2017年3期)2017-04-08
科技视界(2016年26期)2016-12-17
