
基于卷积神经网络的染色体交互预测算法
2020-04-08 09:30马磊磊
电脑知识与技术 2020年3期
马磊磊
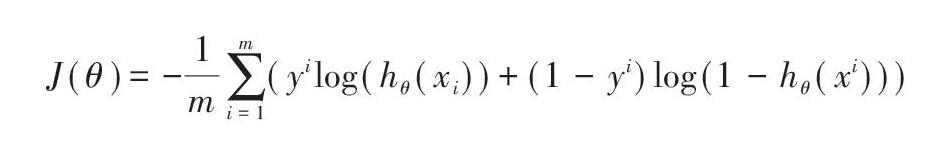
摘要:染色体是由核苷酸序列排列而成,并通过折叠盘旋形成一定的空间结构。染色体在折叠盘旋时相互接触形成调控因子,调控生物体的各项生命活动。现阶段染色体空间结构的表示方法是将三维的空间结构表示成二维的接触矩阵,接触矩阵中的值使用Hi-C等高通量测序技术得到,这个值表示两个染色体片段的接触次数。而生物方法存在实验周期长、代价高等问题,本文针对生物实验中存在的问题,提出了一种基于卷积神经网络的染色体交互预测算法,并取得了较好的预测效果。
关键词:染色体;空间结构;Hi-C;卷积神经网络;染色体交互预测
中图分类号:TP3-05 文献标识码:A
文章编号:1009-3044(2020)03-0198-02
1 研究背景与现状
高通量测序技术引导的生命科学大数据时代的来临,生物信息学相关数据出现爆炸式增长,每天产生TB级别甚至更多的序列数据,对于这些数据的挖掘和分析已经发展成为热点的研究问题。
Hi-C技术[1]是一种以细胞核为研究对象,利用高通量测序,结合生物信息[2]分析方法,研究全基因组范围内整个染色质DNA在空间位置上的关系,并获得高分辨率的染色体调控元件相互作用图谱的技术。Hi-C技术为研究染色体空间结构提供了重要的数据基础。
Hi-C技术本质上是一种高通量测序的生物实验方法[3],这种方法获取染色体交互数据时,存在实验周期长、代价高、误差大等问题。所以使用基于计算的方法挖掘和分析相关数据,进而得到染色体交互数据具有实际意义。
现阶段基于计算的染色体交互数据获取方法主要分为两大类:第一类基于生物实验的Hi-C数据去偏差方法[4]。其主要通过统计计算的方法去除生物实验中存在的偏差,得到较为精确的染色体交互数据。但是这类方法存在和生物实验同样的问题。第二类使用表观修饰数据预测特定区域染色体交互[5]。其主要通过编码表观修饰数据,使用机器学习或者深度学习方法提取数据特征,预测特定区域染色体交互。这类方法可以较快地获取特定区域的染色体交互,但是数据处理流程复杂、预测准确率较低,同时不能获得所有片段的染色体交互数据。
本文针对使用表观修饰数据预测特定区域染色体交互的方法存在的预测准确率低、预测区域具有限制性、数据处理流程复杂等问题,提出了一种以染色体亲水性数据和DNA序列数据作为输入数据,使用卷积神经网络[6]预测全基因组染色体交互的方法。
2 实验分析
本文以染色体亲水性数据,DNA序列数据为原始的输人数据,同时使用Hi-C实验数据作为模型的监督数据[7]。经过数据预处理,数据编码,特征提取等过程,进而预测全基因组染色体交互的可能性。
2.1 算法流程
本文提出的基于卷积神经网络的染色体交互预测模型,主要使用卷积提取编码数据的特征并用于结果预测。基于卷积神经网络预测染色体交互方法的流程主要包括:数据预处理,数据编码,模型设计,实验分析四个关键环节。
数据预处理过程主要收集和划分了染色体亲水性数据,DNA序列数据,同时对Hi-C数据进行了数据转化,其目的是为了生成模型预测的标签数据。最后使用降采样技术,对Hi-C数据生成的标签进行了正負样本平衡处理。
在数据编码阶段,对按照lkb长度划分的染色体亲水性数据和DNA序列数据进行了one-hot编码。由于染色体亲水性数据经过预处理转换成8个类别,分别是0-7,所以对其进行one-hot编码后的结果为8*1000矩阵;DNA序列数据只包含A、G、C、T四个类别,所以对其编码后的结果为4*1000的矩阵。
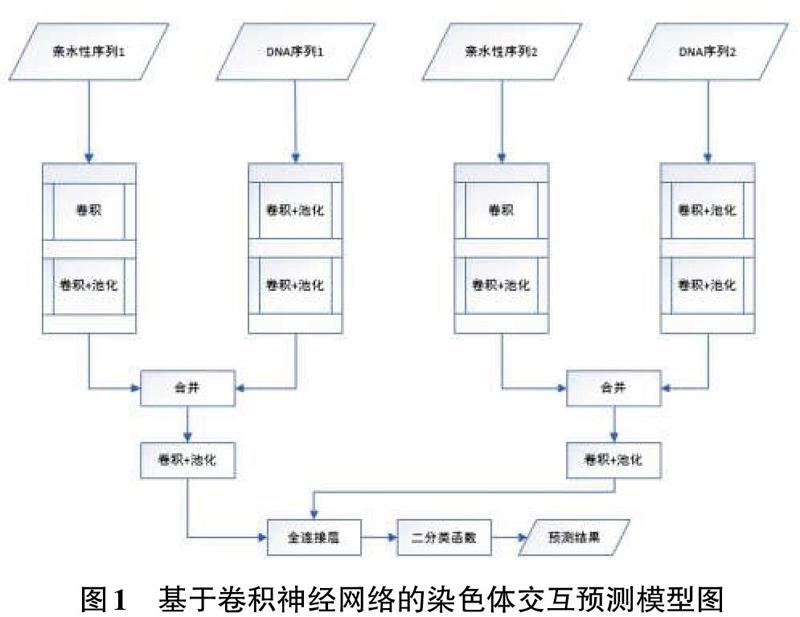
模型设计阶段主要完成了基于卷积神经网络预测染色体交互的模型设计工作。模型分别使用两个卷积网络独立的提取编码后的染色体亲水性数据和DNA序列数据的数据特征,对于提取的特征进行合并,同时再使用卷积神经网络提取更高维度的特征,最后使用全连接层加Softmax函数进行染色体交互可能性预测。
实验分析阶段主要完成了模型训练和预测工作,同时对实验结果进行了简单的分析。
2.2 模型设计
卷积神经网络具有学具表征,特征提取的能力,同时具有尺度不变性。基于卷积神经网络预测染色体交互算法,其模型主要功能模块包括:卷积层,最大池化层,合并层,全连接层及二分类预测层。模型的结构图如图1所示。
2.3 模型训练
基于上述的染色体交互预测模型,我们使用GM12878细胞系数据作为实验的数据集,通过降采样方法,采样了100W对有染色体交互的样本记为正样本,IOOW对没有染色体交互的样本作为负样本。将采样得到的200W对数据,按照9:1的比例划分为训练集和测试集,训练集180W对,测试集20W对。
由于模型是一个二分类模型,模型的正样本标签为l,负样本标签为0。在模型训练过程中,文中选取了交叉熵损失函数[8],使用梯度下降技术进行训练,同时使用Adam算子进行优化。模型的损失函数如下所示:
2.4 模型预测及结果分析
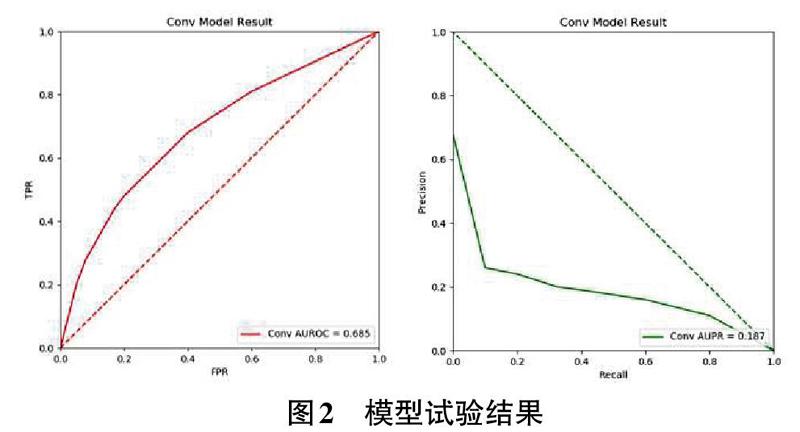
基于上述的训练模型,我们使用20W对测试集对模型训练效果进行了测试和评价。考虑到模型是一个二分类模型,文中使用了AUROC和AUPR作为模型的评价指标。AUROC指ROC曲线下的面积,其意义表示随机获取一个正样本和一个负样本,模型预测为正的结果把正样本排在负样本之前的概率,可以有效地衡量一个二分类模型的效果。AUPR指Recall值和preclsion值形成的曲线下面积。我们使用CM12878细胞系测试数据集的测试结果如图2。
观察模型的预测结果,我们发现,本文提出的基于卷积神经网络的染色体交互预测模型在进行染色体交互可能性预测时AUC值可以达到0.685,同时预测结果的PR曲线下面积也可以达到0.187。
3 結论
本文分析了染色体交互可能的形成机制[9],以染色体亲水性数据,DNA序列数据作为模型的基础输入数据,使用基于卷积神经网络的深度学习模型预测了染色体交互的可能性。实验结果表明,基于卷积神经网络的染色体交互预测模型可以有效地预测全基因组范围内lkb长度的染色体交互,同时可以得到较高的AUC值,本模型对于使用深度网络模型预测染色体交互具有很好的实用价值和参考意义。
参考文献:
[1] JinF, Li Y.Dixon J R.et al.A high-resolution map of thethree-dimensional chromatin interactome in human cells[J].Nature, 2009,503(7475):290-294.
[2]董建成霍奇曼,林安华,生物信息学[M].北京:科学出版社,2010.
[3]吕红强,郝乐乐,刘源,等.基于生物信息学的Hi-C研究现状与发展趋势(三维基因组专刊稿件)[J].遗传,2019: 0-0.
[4] Lettice L A.Disruption of a long-range cis-acting regulatorfor Shh causes preaxial polydactyly[J]. Proc. Natl Acad. Sci.USA 99, 2002: 7548-7553.
[5] Wenran L,Hung W W, Rui J.DeepTACT: predicting 3D chro-matin contacts via bootstrapping deep learning[J]. Nucleic Ac-ids Research. 2018 (10):10.
[6] S.Chopra, R.Hadsell, and Y.LeCun. Leaming a similarity met-ric discriminatively, with application to face verification, inProc[J]. IEEE Comput. Soc. Conf. CVPR, 2002:539-546.
[7] Rao,S.S.et al.A 3D map of the human genome at kilobaseresolution reveals principles of chromatin looping[J]. Cell. 2014 (159):1665-1680.
[8]阎平凡,张长水.人工神经网络与模拟进化计算[M].北京:清华大学出版社,1900.
[9]吴燕如,珠杰,管美静,基于神经网络的目标检测技术研究综述及应用[J].电脑知识与技术,2019,15(33):181-184.
猜你喜欢
阿来研究(2020年1期)2020-10-28
科学之谜(2019年3期)2019-03-28
科学之谜(2018年8期)2018-09-29
计算机应用(2016年12期)2017-01-13
新世纪水泥导报(2016年1期)2016-07-01
中央社会主义学院学报(2016年2期)2016-05-04
中央民族大学学报(自然科学版)(2015年2期)2015-06-09
土木建筑工程信息技术(2013年1期)2013-10-17
