
恩曲他滨的合成工艺优化
2020-06-27 08:20顾继山吴晓宇孙国锋丁一刚刘生鹏
武汉工程大学学报 2020年2期
李 苏,顾继山,吴晓宇,熊 芸,孙国锋,丁一刚,刘生鹏
武汉工程大学化工与制药学院,湖北 武汉 430205
恩曲他滨(emtricitabine,FTC),化学名为4-氨基-5-氟-1-[(2R,5S)-2-羟甲基-1,3-氧硫杂环戊烷-5-基]-2(1H)-嘧啶酮,是一种核苷类逆转录酶抑制剂。该药由美国Gilead 公司开发,于2003 年经美国FDA 批准上市,用于治疗由人类免疫缺陷病毒(HIV)感染引起的艾滋病,以及乙型肝炎病毒(HBV)引起的乙型肝炎[1]。自上市以来,临床证明FTC 的抗病毒失败率低,治疗效果好,是治疗艾滋病和乙型肝炎最成功的药物之一[2-3]。
传统的FTC 滨合成路线主要采用化学合成的方法[4-6],总收率均在29.8%~51%之间,且都存在成本高、反应步骤多、毒性大和污染环境等缺点[7-8]。目前,由于脂肪酶具有高度的立体选择性、热稳定性、底物广泛性、催化活性、环境友好性和回收利用性等[9-11],而被广泛用于药物的合成中[12-14]。本文在参考已报道的工艺基础上[8,12,15],以苯甲酸钠2 和溴乙醛缩二乙醇3 为原料,缩合得到苯甲酰氧基乙醛缩二乙醇4,将4 在浓度为2.4 mol/L 盐酸溶液中水解得到苯甲酰氧基乙醛5,5 与1,4-二噻烷-2,5-二醇6 在脂肪酶Novozyme 435 催化下得到(2R,5S)-2-苯甲酰氧甲基-5-乙酰氧基-l,3-氧硫杂环戊烷7,7 与硅烷化5-氟胞嘧啶8 进行偶联得到4-氨基-5-氟-1-[(2R,5S)-2-苯甲酰氧甲基-1,3-氧硫杂环戊烷-5-基]-2(1H)-嘧啶酮9,将其水解后可得终产物FTC(图1),总收率为51.3%,FTC 纯度为99.7%。
1 实验部分
1.1 仪器与试剂
Bruker Avance NEO-600 MHz 型核磁共振波谱仪(德国Bruker 科技有限公司);SHIMADZU CT0-20AC 高效液相色谱仪,SHIMADZU-2014 气相色谱仪(日本岛津仪器公司);WRS-3A 熔点仪(上海仪电物理光学仪器有限公司);JASCO P-1030 自动旋光仪(日本分光株式会社)。
苯甲酸钠,1,4-二噻烷-2,5-二醇,5-氟胞嘧啶(阿拉丁生化科技股份有限公司);溴乙醛缩二乙醇,三乙烯二胺,六甲基二硅胺烷(武汉格奥化学技术有限公司);盐酸,乙酸苯酯,正己烷,无水乙醇,三氟乙酸(上海麦克林生化科技有限公司);四氢呋喃,N,N-二甲基甲酰胺,乙酸乙酯,二氯甲烷,三乙胺,甲醇等,(国药集团化学试剂有限公司);所有试剂均为分析纯。Novozyme 435(诺维信公司)。
1.2 结果检测
色谱柱:Chiralpak AD-H 柱(250 mm×4.6 nm,5 μm);流动相:V(正己烷)∶V(无水乙醇)∶V(甲醇)∶V(三氟乙酸)∶V(三乙胺)=800∶150∶50∶1∶1;柱温:40 ℃;检测器波长280 nm;流速设定为1.0 mL/min,进样量20 μL。精密称取合成的FTC约5.0 mg,加入1 mL 甲醇溶解,转移至10 mL 容量瓶中,并用流动相稀释至刻度,制成浓度约为0.5 mg/mL 的溶液。结果得出目标产物恩曲他滨1的纯度为99.7%,符合中国药典标准。
1.3 实验方法
1.3.1 苯甲酰氧基乙醛缩二乙醇(4)的合成 称取化合物2(9.925 g,68 mmol)于1 L 三口烧瓶中,并加入N,N-二甲基甲酰胺(N,N-Dimethyl formamide,DMF,300 mL),加入3(19 mL,124.9 mmol),搅拌溶解,于135 ℃回流反应2 h 后,再加入化合物2(9.925 g,68 mmol)继续反应14 h,TLC 监测3消失。反应完毕后,旋蒸除去DMF,加入乙酸乙酯溶解,分别用蒸馏水和饱和氯化钠溶液洗涤,萃取有机层,无水硫酸钠干燥,旋蒸除去溶剂,得到红色粘稠液体26.25 g,收率88.2%。1H NMR(600 MHz,DMSO-d6),δ:8.07(d,2H,J=8.0 Hz,Ar-H),7.58(t,1H,J=7.4 Hz,Ar-H),7.45(t,2H,J=7.7 Hz,Ar-H),4.85(t,1H,J=5.5 Hz,-CH),4.36(d,2H,J=5.5 Hz,-CH2),3.71(q,4H,-CH2),1.23(t,6H,J=7.1 Hz,-CH3)。13C NMR(600 MHz,DMSO-d6),δ:166.27,133.06,130.04,129.73,128.39,99.75,64.47,62.54,15.36。与文 献[8]一致。

图1 FTC 的合成路线Fig.1 Synthesis route of FTC
1.3.2 苯甲酰氧基乙醛(5)的合成 称取化合物4(3.0 g,12.6 mmol)、20 mL 四氢呋喃于50 mL 三口烧瓶中,升温至60 ℃回流反应,磁力搅拌下,滴加入2.4 mol/L(10 mL)的盐酸。1 h 后,结束反应。旋蒸除去四氢呋喃,加入乙酸乙酯溶解,用饱和碳酸氢钠溶液和氯化钠溶液洗涤,萃取有机层,无水硫酸钠干燥,旋蒸除去溶剂。粗品经层析柱纯化,得到浅黄色油状液体1.77 g,收率为84.7%。1H NMR(600 MHz,DMSO-d6),δ:9.73(s,1H,-CHO),7.96~8.17(d,2H,J=7.45 Hz,Ar-H),7.61(d,1H,J=7.2 Hz,Ar-H),7.48(d,2H,J=7.7 Hz,Ar-H),4.91(s,2H,-CH2)。13C NMR(600 MHz,DMSO-d6),δ:196.11,166.21,133.86,130.15,129.14,128.78,69.26。与文献[8]一致。
1.3.3 (2R,5S)-2-苯甲酰氧甲基-5-乙酰氧基-1,3-氧硫杂环戊烷(7)的合成 称取化合物5(0.26 g,1.58 mmol)、6(0.14 g,0.95 mmol)、乙酸苯酯(0.6 mL,4.74 mmol)、三乙胺(0.22 mL,1.43 mmol)、DABCO(0.013 g,0.094 8 mmol,0.1 eq)溶于10 mL 无水四氢呋喃中,40 ℃回流反应6 h。向反应混合物中加入0.38 g 0.4 nm 分子筛,缓慢搅拌1 h,加入磷酸盐缓冲溶液(pH=7.4,3 mL),250 mg(25 mg/mL)Novozyme 435 继续搅拌8 h 后停止。过滤反应混合物,滤液用饱和氯化钠溶液洗涤,萃取有机层,无水硫酸钠干燥,旋蒸除去溶剂。粗品经层析柱纯化,得到无色油状液体0.371 g,收率为82.1%,ee值为99%(文献[8]为82%)。1H NMR(600 MHz,DMSO-d6),δ:8.05(d,2H,J=8.0 Hz,Ar-H),7.55(d,1H,J=7.2 Hz,Ar-H),7.43(d,2H,J=7.5 Hz,Ar-H),5.43(t,1H,J=4.7 Hz,-CH),4.75~4.94(m,1H,-CH),4.01~4.57(m,2H,-CH2),2.97~3.24(m,2H,-CH2),2.04(s,3H,-CH3)。13C NMR(600 MHz,CDCl3),δ:171.31,166.01,133.21,129.75,128.44,100.06,81.10,66.42,38.69。与文献[12]一致。
1.3.4 4-氨基-5-氟-1-[(2R,5S)-2-苯甲酰氧甲基-1,3-氧硫杂环戊烷-5-基]-2(1H)-嘧啶酮(9)的制备 称取5-氟胞嘧啶(0.52 g,4.0 mmol)、3 mL 六甲基二硅胺烷、0.1 g 催化剂硫酸铵、15 mL DMF 于100 mL 三口烧瓶中。升温至100 ℃,回流反应。反应至反应液澄清透明,停止反应并旋蒸除去六甲基二硅胺烷,冷却至室温,得到化合物8。加入1.4 mL 三乙胺混合均匀以备用。
将化合物7(0.3 g,1.0 mmol)溶于6 mL 无水二氯甲烷中,倒入上述制备的8,并加入0.3 g 孔径0.4 nm 分子筛,于80 ℃下回流搅拌反应8 h。反应结束后,加入乙酸乙酯溶解,用饱和碳酸氢钠溶液、蒸馏水和盐水洗涤,萃取有机层,无水硫酸钠干燥,旋蒸除去溶剂,用乙醇重结晶,得到粗产物混合物,为浅黄色粉末。不经纯化,直接进入下一步。1.3.5 FTC(1)的合成 取化合物9(0.3 g,0.7 mmol)溶于50 mL 甲醇和50 mL 氨水溶液中混合,在室温下搅拌10 h 后,抽滤,旋蒸除去溶剂,粗品经层析柱纯化,得到0.15 g 白色粉末,收率为83.6%,纯度为99.7%,ee 值为99.5%。旋光度[α]25D=-119.4°(c 1.05,MeOH)[文 献[4]为-116°,c 1.05,MeOH];m.p.180~185 ℃;1H NMR(600 MHz,DMSO-d6),δ:8.19(d,1H,J=7.2 Hz,-CH),7.81(s,1H,NH),7.57(s,1H,NH),6.13(s,1H,NH),5.41(t,J=5.54 Hz,1H,-OH),5.17(t,1H,J=3.8 Hz,-CH),3.65~3.82(m,2H,-CH2),3.39(dd,1H,J=24.5 Hz,-CH2),3.12(dd,1H,J=11.9 Hz,4.3 Hz,-CH2)。13C NMR(600 MHz,DMSO-d6),δ:157.95,153.45,137.06,135.47,126.26,87.01,62.60,37.24。与文献[7]一致。
2 结果与讨论
2.1 化合物5 反应条件优化
考察了以88%甲酸水溶液、不同浓度盐酸和对甲苯磺酸等作为H+供体时的反应条件(表1)。可以得出,当反应温度由室温升至60 ℃时,副产物乙醇蒸发,使反应向正反应方向进行。并且,采用2.4 mol/L 盐酸时,得到最高收率,88.2%。

表1 用于缩醛水解反应的不同催化剂的反应条件和结果*Tab.1 Reaction conditions and results over different catalysts used for hydrolysis reaction
2.2 脂肪酶催化合成化合物7 的优化
2.2.1 溶剂对酶催化反应的影响 酶促催化反应中,酶的催化活性和立体选择性会受到溶剂的logP 值和极性的影响,因此必须选择合适的有机溶剂。选取除水处理后的甲苯、二氯甲烷、四氢呋喃、乙腈、甲醇和N,N-二甲基甲酰胺6 种有机溶剂进行对比。从表2 可以看出,四氢呋喃作为溶剂时,转化率和对映体选择性最高。在强亲水性溶剂(logP <0)中,反应的转化率很低,是因为亲水性溶剂导致Novozyme 435 脂肪酶周围必需的水层被剥离,脂肪酶的结构被破坏,催化活性降低。而非极性溶剂(logP >2.5)的疏水性虽然能使酶结构稳定,但是其对映体选择性很低。
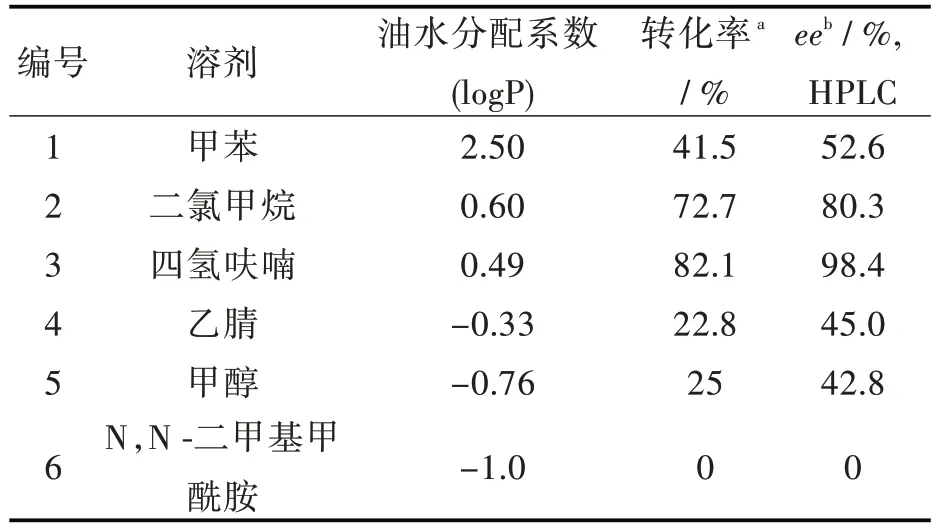
表2 溶剂对酶催化反应的影响Tab.2 Effect of solvent on reaction of lipase-catalysis
2.2.2 温度对催化反应的影响 由于脂肪酶的本质是蛋白质,温度会影响酶的活性、稳定性和对映体选择性,所以必须考察温度对脂肪酶的催化反应的影响。通常情况下,随着温度升高,反应速度也随之升高。当升高到一定温度后,酶逐渐变性,失去催化活性。本研究考察了25~70 ℃之间的影响,结果如表3 所示。可以看到,转化率在40 ℃时达到最大值,因此选取40 ℃为最佳反应温度。

表3 温度对酶催化反应的影响Tab.3 Effect of temperature on lipase-catalysis reaction
3 结 论
本文优化了FTC 的合成工艺路线。优化了苯甲酰氧基乙醛5 的合成工艺;以及在关键中间体7的合成过程中,选取Novozyme 435 脂肪酶为催化脂肪酶用于催化反应;在催化过程中确定了最佳反应溶剂和反应温度,分别为四氢呋喃和40 ℃,得到ee 值>99%,收率为82.1%的化合物7。经过优化后,得到总收率为51.3%,纯度为99.7%的FTC。综上所述,本文的研究内容为其他不对称手性化合物的合成提供了一种有参考价值的思路和方案。
猜你喜欢
工业催化(2022年9期)2022-10-17
中华胰腺病杂志(2022年4期)2022-08-23
食品安全导刊(2021年20期)2021-08-30
求知导刊(2019年15期)2019-08-30
广西科技大学学报(2018年2期)2018-09-10
中小学实验与装备(2016年1期)2016-04-19
分析化学(2015年7期)2015-07-30
安徽化工(2015年2期)2015-06-29
绿色科技(2015年12期)2015-01-16
中学教学参考·理科版(2014年4期)2014-08-21
