
气相色谱法测定水果中咪鲜胺残留量的前处理方法探究
2020-07-18 04:06刘志鹏杨李胜黄敏兴柯华南
食品工业 2020年6期
刘志鹏,杨李胜*,黄敏兴,柯华南
1. 广东省生物工程研究所/广州甘蔗糖业研究所(广州 510000);2. 中国轻工业甘蔗制糖工程技术研究中心(广州 510000)
咪鲜胺又名施保克,其通用名为Prochloraz,化学名为N-丙基N-[2-(2, 4, 6-三氯苯氧基)乙基]-1咪唑基甲酰胺(LPAC),在动植物体内的最终代谢产物为2, 4, 6-trichbrophenbor(2, 4, 6-TCP)[1]。咪鲜胺及其制剂进入我国市场后,推广应用发展很快,已经被广泛应用于多种农林产品的生产、贮存和运输等过程中[2]。咪鲜胺是一种咪唑类广谱杀菌剂,可用于谷类、热带和亚热带水果、蔬菜等经济作物的病虫害的防治[3]。尽管咪鲜胺是低毒杀菌剂,但其最终产物2,4, 6-三氯苯酚是环境中的重要污染物之一,会污染生态环境,并会对人类健康造成潜在威胁。因此,其在水果生产和储藏中的残留问题越来越受到人们关注[4]。咪鲜胺最大残留限量被美国和欧盟严格监测,在水果中的最大残留限量(MRL)一直在很低的水平上[5]。
目前,国内外已发表的对咪鲜胺残留物的检测主要采用色谱法[6],主要是气相色谱-质谱联用[7]、高效液相色谱-质谱、高效液相色谱和气相色谱。尹桂豪等[8]采用气相色谱法对芒果中咪鲜胺残留分析;韩丽君等[9]采用GC-ECD研究了咪鲜胺及其代谢物在水稻中的总残留量检测方法;De-Paoli等[10]对咪鲜胺及其代谢产物残留在蔬菜、水果和小麦样品组合简化测定气相色谱法。
但这些方法却存在着一些问题:一是气相色谱-质谱联用和高效液相色谱-质谱的前处理方法虽然简单,但仪器昂贵且难以普及应用;二是使用高效液相色谱只可以检测咪鲜胺的残留量,但不能检测其全部的代谢物,尤其是2, 4, 6-三氯苯酚的残留量,且灵敏度比气相色谱低[4];三是样品前处理方法技术落后,在处理大量样品时需要消耗大量的试剂、玻璃仪器和时间,效率较低。
此次试验同样采用气相色谱法对水果中的咪鲜胺及其代谢产物残留检测方法进行研究,对样品前处理方法进行优化,缩短提取过程的时间也能最大限度地提取咪鲜胺及其代谢产物;减少多余的玻璃仪器使用;并且采用一种新的方法进行净化,避免了使用浓硫酸带来的安全性隐患以及在操作过程中调节pH带来的不确定性,提高了方法的选择性和灵敏度,从而建立一套快速、便捷、安全、清洁、耗时短、低成本且灵敏度高的检测方法。
1 材料与方法
1.1 材料与仪器
试样苹果、葡萄、西瓜、柑橘等水果,市售。
咪鲜胺、2, 4, 6-三氯苯酚(纯度均不少于99.0%),德国DR公司;吡啶盐酸盐、盐酸、浓硫酸、无水硫酸钠(使用前在55 ℃下烘干2 h,冷却后使用),均为分析纯,广州化学试剂厂;二氯甲烷、丙酮、石油醚、乙腈、正己烷,均为色谱级,美国默克公司;试验用水均为一级水。
7890B气相色谱仪,美国安捷伦公司;TLE204E电子天平,梅特勒-托利多仪器(上海)有限公司;DK-2可调式电砂浴锅,北京市永光明医疗仪器有限公司;RV 8V旋转蒸发仪,艾卡(广州)仪器设备有限公司(德国IKA);SYG-1230水浴箱,上海珂淮仪器有限公司;T25均质机,艾卡(广州)仪器设备有限公司(德国IKA)。
1.2 方法
1.2.1 标准溶液配制
咪鲜胺标准工作液:称取10 mg咪鲜胺标准物质,用丙酮溶解并定容至100 mL,配制成质量浓度为100 mg/L的标准溶液储备液,用丙酮再次稀释配制成质量浓度为1.0 mg/L的咪鲜胺标准工作液。
2, 4, 6-三氯苯酚工作液:称取10 mg 2, 4, 6-三氯苯酚标准物质,用丙酮溶解并定容至100 mL,配制成质量浓度为100 mg/L的标准溶液储备液,用丙酮再次稀释配制成质量浓度为1.0 mg/L的2, 4, 6-三氯苯酚标准工作液。
1.2.2 水果储备基质液
分别称取5份40 g苹果试样于5个100 mL的平底烧杯中,各加入50 mL的乙腈,匀浆机高速匀浆2 min,过滤,收集于装有5 g氯化钠的100 mL具塞比色管中,盖上盖子,剧烈振荡1 min,静置30 min,从比色管中吸取上层溶液于250 mL的平底烧杯中,摇匀,备用。
1.2.3 GC色谱条件
检测方法采用GC-ECD法,色谱柱采用DB-5(30 m×0.320 mm×0.25 μm)的弹性石英毛细管柱。仪器检测条件:柱温70 ℃以40 ℃/min的速度升温到245℃(保留5 min);进样口温度260 ℃;ECD温度300℃;分流进样,分流比40∶1;色谱柱的载气气体为氮气,流量2.5 mL/min;进样量1.0 μL。
1.2.4 标准曲线制作
准确吸取0.25,0.50,1.0,2.0和3.0 mL咪鲜胺标准工作液于水解试管中,氮吹浓缩近干,加入5 g吡啶盐酸盐,水解方法、净化方法按照最优方法进行,待测。
2 结果与分析
2.1 对前处理净化方式优化分析
分3组,每组各取6个50 mL平底烧杯并分别加入10 mL储备基质液,然后在烧杯中分别加入1.0 mL 1.0 mg/L的2, 4, 6-三氯苯酚工作液(模拟咪鲜胺已完全水解),在振荡器上混匀,放在氮吹仪上浓缩近干,待用。
1) 硫酸的净化方法:将上述烧杯用20 mL正己烷分数次冲洗并全部转移到分液漏斗中,加入5 mL硫酸,振荡1 min,静止分层后,弃去硫酸,重复3次。然后用蒸馏水洗涤有机相中的残留硫酸,洗涤至中性,收集有机相,经无水硫酸钠脱水干燥后浓缩低于5 mL,并用正己烷定容至5 mL,上机测定,结果如图1所示。

图1 利用浓硫酸进行净化的咪鲜胺残留量色谱图
2) PSA 100 mg+C18100 mg+MgSO4300 mg QuECh-ERS净化管的净化方法:将上述烧杯用正己烷分数次洗涤烧杯并全部转移到5 mL容量瓶,并定容至5 mL。然后全部转移到装有PSA 100 mg+C18100 mg+MgSO4300 mg的QuEChERS净化管中净化,上机测定,结果如图2所示。
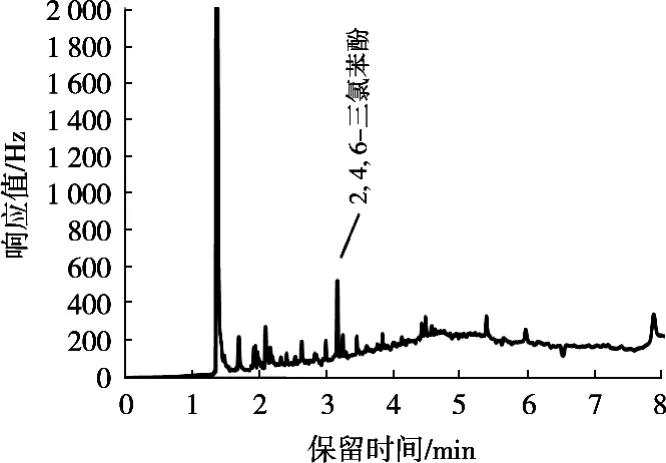
图2 利用净化管进行净化的咪鲜胺残留量色谱图
3) 弗罗里硅土柱的净化方法:将上述烧杯分数次加入5 mL正己烷,并全部转移到弗罗里硅土柱上(已用洗脱液活化),弃去滤液,用25 mL丙酮-正己烷(90∶10,V/V)洗脱溶剂洗脱,用25 mL带刻度试管接收滤液,氮吹浓缩低于5 mL,最后用正己烷定容至5 mL,上机测定,结果如图3所示。

图3 用弗罗里硅土柱进行净化的咪鲜胺残留量色谱图
经计算,2, 4, 6-三氯苯酚回收率得到的结果如表1所示。由图谱和回收率计算结果可知,QuEChERS净化管的填料对样品中2, 4, 6-三氯苯酚的吸附作用很强,能把试样中大部分2, 4, 6-三氯苯酚吸附在净化管中,并且对于其他杂峰的净化效果不显著,甚至是没有得到净化效果,影响咪鲜胺最后的定量计算,故排除使用QuEChERS净化管的使用;而浓硫酸和弗罗里硅土柱两种的净化效果能达到检测要求,回收率均在90%以上,但净化过程使用浓硫酸,容易发生危险,而弗罗里硅土柱净化则使用过程相对简单,不会对实验人员造成潜在的危险以及在之后调节pH的不确定性也可以避免。故使用弗罗里硅土柱作为净化的装置。
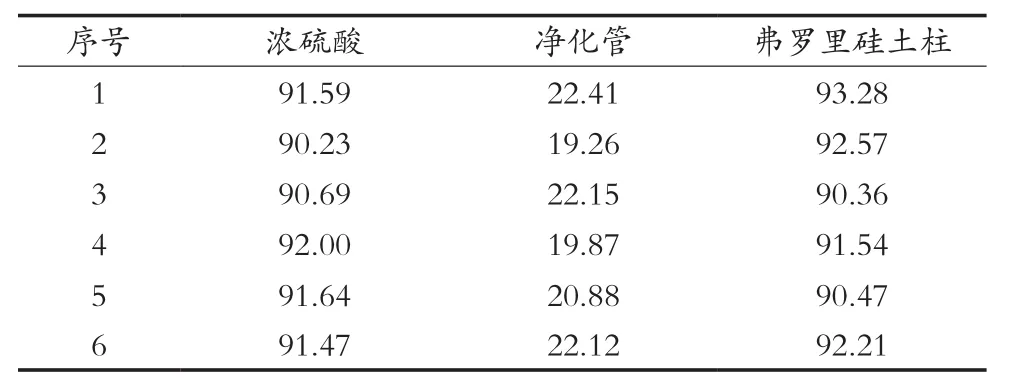
表1 不同净化方法下的2, 4, 6-三氯苯酚回收率 %
2.2 对洗脱溶剂优化分析
分3组,每组各取6个50 mL平底烧杯并分别加入10 mL储备基质液,然后在烧杯中分别加入1.0 mL 1.0 mg/L的2, 4, 6-三氯苯酚工作液(模拟咪鲜胺已完全水解),在振荡器上混匀,放在氮吹仪上浓缩近干,加入5 mL正己烷,盖上蜡膜,待用。
根据2.1优化,将弗罗里硅土柱用5 mL表2中不同配比洗脱液进行条件活化,当溶剂液面到达柱吸附层表面时,马上倒入上述样品溶液,用25 mL的具刻度试管收集洗脱液,分别用25 mL相同配比的溶液冲洗烧杯后洗脱净化柱。将装有洗脱液的试管置于氮吹仪,在50 ℃水浴中浓缩近干,用正己烷准确定容至5 mL,混匀,待测,结果如表2所示。
结果显示,在丙酮与正己烷的配比是95∶5(V/V)和90∶10(V/V)及丙酮与二氯甲烷的配比是95∶5(V/V)时,2, 4, 6-三氯苯酚几乎能全部被洗脱下来,其回收率均在90%以上,均满足对检测的要求。但是95∶5(V/V)的丙酮+二氯甲烷同时会对杂质的回收率也明显增加,特别对咪鲜胺临近杂峰的洗脱,此杂峰会影响咪鲜胺的的定量检测,而在95∶5(V/V)和90∶10(V/V)的丙酮+正己烷洗脱溶剂中,由于丙酮价格比正己烷的高,为降低检测成本,选择90∶10(V/V)的丙酮+正己烷相对合适。
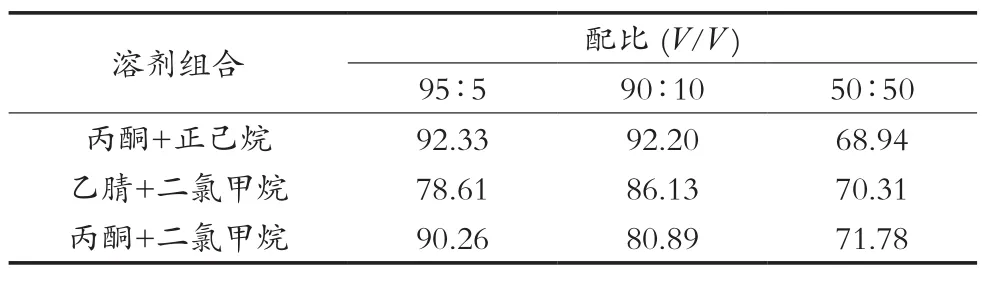
表2 不同洗脱溶剂的配比下2, 4, 6-三氯苯酚平均回收率 %
2.3 对水解装置优化分析
分3组,每组各取6支带具塞刻度的25 mL水解试管,加入1.0 mL咪鲜胺标准工作液,氮吹浓缩近干,加入5 g吡啶盐酸盐,按照表3中的水解条件进行水解,冷却至室温后,加入适量的水溶解吡啶盐酸盐,并全部转移到250 mL的分液漏斗中,试管用50 mL水分数次冲洗并转移到分液漏斗中,石油醚萃取2次(每次50 mL),弃去水相,合并有机相;浓缩近干,净化方法按照2.1、2.2优化出的最优方法进行。
经多次反复试验,在沙浴与25 mL的水解试管装置下,一个砂浴锅能同时加热20~30个试样,同时进行水解,完全可以进行大批量处理样品。水解过程并没有沸腾现象,安全可靠。同时,反应后的试管容易清洗,能大大增加工作效率。其余方式操作繁琐,不利于检测多个样品,不能提高处理效率。

表3 水解装置及条件表
2.4 对提取溶液优化分析
分3组,分别称取40 g苹果试样于250 mL的平底烧杯中,加入1 mL 1.0 mg/L的咪鲜胺标准工作液,按照表5设计对应加入各种用量的溶剂,并在匀浆机上高速匀浆2 min,过滤,收集滤液于装有5 g氯化钠的100 mL具塞试管中,盖上盖子,剧烈振荡1 min,在室温下静置30 min,使之分层,吸取20 mL上层溶液于25 mL的带刻度的试管中,氮吹仪上浓缩近干;加入5 g吡啶盐酸盐,水解方法使用按照2.3筛选出的最佳水解装置进行水解,水解1.5 h;净化方法按照2.1、2.2优化出的最优方法进行,上机待测,咪鲜胺回收率结果见表4。
NY/T 1456—2007中前处理提取液是用80 mL丙酮,并且需要浸泡过夜后仍需要振荡60 min,试验时间长,不便于检测短时高效的要求。上述结果表明丙酮和乙腈在适当的体积中均能满足检测要求,而乙腈的提取回收率均在90%以上,溶剂用量各水平差异不明显。故使用乙腈作为提取溶剂,其用量为50 mL。
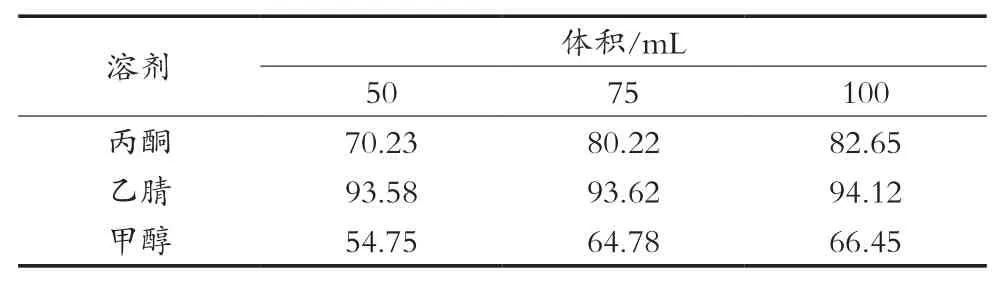
表4 不同提取溶剂试验下咪鲜胺平均回收率 %
2.5 方法的线性范围和检出限
将1.2.4配制好的咪鲜胺标准曲线按照上述优化后的条件进行处理进样,以质量浓度(mg/L)为横坐标,峰面积为纵坐标,绘制标准曲线(见图4)。线性方程为y=12 991.72x-118.34,其相关性r=0.999 53,这说明在0.05~0.6 mg/L线性范围内有良好的线性关系。通过实际低浓度添加试验,当信噪比等于3时,咪鲜胺的检出限为0.005 mg/kg,满足试验要求。

图4 咪鲜胺标准工作曲线
2.6 方法的回收率和精密度
按照上述优化后的条件,对苹果、葡萄、西瓜、柑橘4种样品分别添加0.05,0.20和0.40 mg/kg三个浓度水平,做加标回收试验,每个浓度6平行,结果见表5。咪鲜胺的平均回收为87.2%~101.1%,相对标准偏差为2.1%~5.5%。
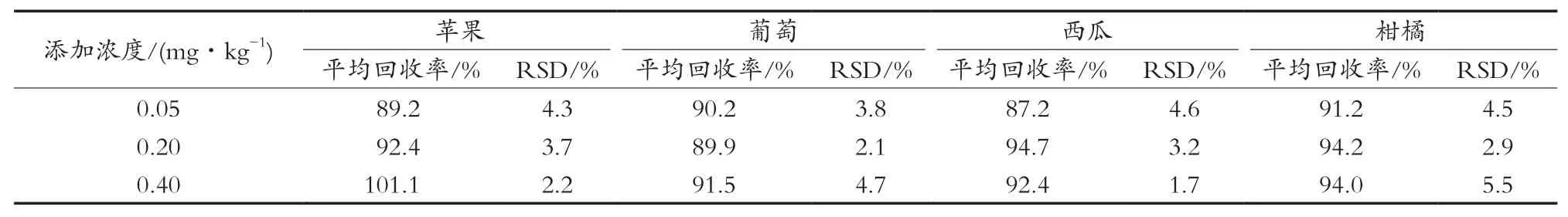
表5 4类蔬果在3个添加水平下的平均回收率和相对标准偏差(n=6)
2.7 方法在实际样品上的应用
在广州市各大市场上随机抽取橙子、桃子、草莓、葡萄、西瓜、苹果等水果,共109批次,按照优化后的试验条件,每批次3平行进行检测。结果显示共21批次检出咪鲜胺及其代谢物,其中葡萄与柑橘类检出率最高。其浓度范围在0.011 1~0.291 mg/kg之间,RSD范围在0.9%~7.2%之间。

图5 葡萄阳性样品色谱图
图6 0.2 mg/L 2, 4, 6-三氯苯酚标准点色谱图
3 结论
此次试验建立在GC-ECD检测器的基础上。经试验验证,测定水果中咪鲜胺残留量的前处理方法优化为:称取40 g试样于250 mL烧杯中,加入50 mL乙腈,高速均质2 min,过滤;收集滤液于装有5 g氯化钠的100 mL具塞试管中,盖上盖子,剧烈振荡1 min,在室温下静置30 min,使之分层,吸取20 mL上层溶液于25 mL的带刻度的水解试管中,氮吹浓缩近干后加入5 g吡啶盐酸盐,置于210~240 ℃的沙浴中水解1.5 h;冷却后,加入10 mL蒸馏水使吡啶盐酸盐溶解,并用50 mL蒸馏水分次冲洗水解管转移到250 mL分液漏斗中,用石油醚萃取2次(每次50 mL),合并有机相;将有机相转移到100 mL烧杯中,浓缩至干,用10 mL正己烷分次洗烧杯,溶液转移到经丙酮-正己烷混合溶液(90∶10,V/V)活化的弗罗里硅土柱,弃去滤液,用25 mL丙酮-正己烷混合溶液(90∶10,V/V)分数次洗脱,收集滤液,氮吹并定容5 mL,供测定。该前处理方法在保证符合检测要求的基础上,简化了实验步骤,优化了实验的条件,能同时检测多个样品,既缩短检测的时间,又节省成本费用,安全可靠,方法简单,且灵敏度高,适用于批量水果样品中咪鲜胺及其代谢物残留的定性和定量分析。
猜你喜欢
发明与创新·小学生(2022年12期)2022-12-09
世界农药(2022年8期)2022-09-02
当代水产(2021年10期)2022-01-12
农药科学与管理(2019年7期)2019-11-29
江西农业大学学报(2018年6期)2019-01-14
食品与机械(2017年5期)2017-07-05
环境科技(2016年4期)2016-11-08
红领巾·探索(2014年7期)2014-10-10
中学生数理化·八年级物理人教版(2014年2期)2014-04-02
河南城建学院学报(2014年4期)2014-02-27
