
一测多评法测定不同产地紫苏子中4种活性成分的含量△
2020-08-21 06:56杨文惠罗宇琴孙冬梅潘礼业魏梅霍文杰陈向东
中国现代中药 2020年6期
杨文惠,罗宇琴,孙冬梅,潘礼业,魏梅,霍文杰,陈向东
广东一方制药有限公司/广东省中药配方颗粒企业重点实验室,广东 佛山 528244
紫苏子为唇形科植物紫苏Perillafrutescens(L.)Britt. 的干燥成熟果实,秋季果实成熟时采收,除去杂质,晒干。紫苏资源在我国分布范围较广,分布于广东、广西、湖北、云南、贵州、四川、台湾、浙江、安徽、福建等省区[1-2],紫苏属植物喜温、短日照,适应能力强。紫苏子的化学成分主要包括黄酮类、挥发性成分、酚酸类、脂肪酸及无机元素等,其中酚酸类成分包括咖啡酸-3-O-葡萄糖苷、咖啡酸、迷迭香酸、迷迭香酸-3-O-葡萄糖苷等,此外还包括黄酮类如木犀草素、芹菜素等,其性温,味辛,具有降气化痰、止咳平喘、润肠通便的功能,临床主要用于痰壅气逆、咳嗽气喘、肠燥便秘等。目前,文献对紫苏子的含量测定研究多集中于对不饱和脂肪油,如α-亚油酸、α-亚麻酸、迷迭香酸及木犀草素等成分,而咖啡酸、芹菜素等的含量测定研究报道较少,而中药多以有效成分群发挥其药用价值,因此,采用一测多评法(QAMS)[3-6]研究有其明显的应用价值。
QAMS是利用中药中有效成分内在函数关系和比例关系,只测定1个有效成分(性质稳定且易得到的对照品)来实现多个成分(性质不稳定且难得到的对照品)的同时测定[3-6],该方法快速、简便并且能实现同时检测多个成分,是一种符合中药多成分特点的多指标质量评价模式,已经广泛应用于中药的质量控制工作当中。
本研究采用高效液相色谱法同时测定紫苏子中咖啡酸、迷迭香酸、木犀草素、芹菜素的含量,并利用上述4种化学成分之间的函数关系和比例关系,建立一测多评方法,选用含量较高的迷迭香酸为内标物质,得到咖啡酸、木犀草素、芹菜素与迷迭香酸之间的相对校正因子(f),计算这各化学成分的含量,为中药材紫苏子多成分含量的测定与质量控制提供了全新的分析模式。
1 材料
1.1 仪器
仪器:Waters超高效液相色谱仪(沃特世公司,H-Class),Waters HSS T3色谱柱(100 mm×2.1 mm,1.8 μm),Agilent SB C18色谱柱(100 mm×2.1 mm,1.8 μm),YMC Triart色谱柱(100 mm×2.1 mm,1.9 μm),万分之一天平(梅特勒-托利多公司,ME204E),百万分之一天平(梅特勒-托利多公司,XP26),电热恒温水浴锅(上海一恒科技有限公司,HWS-28),数控超声波清洗仪(昆山市超声仪器有限公司,KQ500DE),超纯水系统(默克股份有限公司,Milli-Q Direct)。
1.2 试药
本实验所用的紫苏子药材由广东一方制药有限公司提供,经广东一方制药有限公司魏梅主任中药师鉴定为紫苏子P.frutescens(L.)Britt.的干燥成熟果实,样品保存于质量中心,样品采集信息见表1。
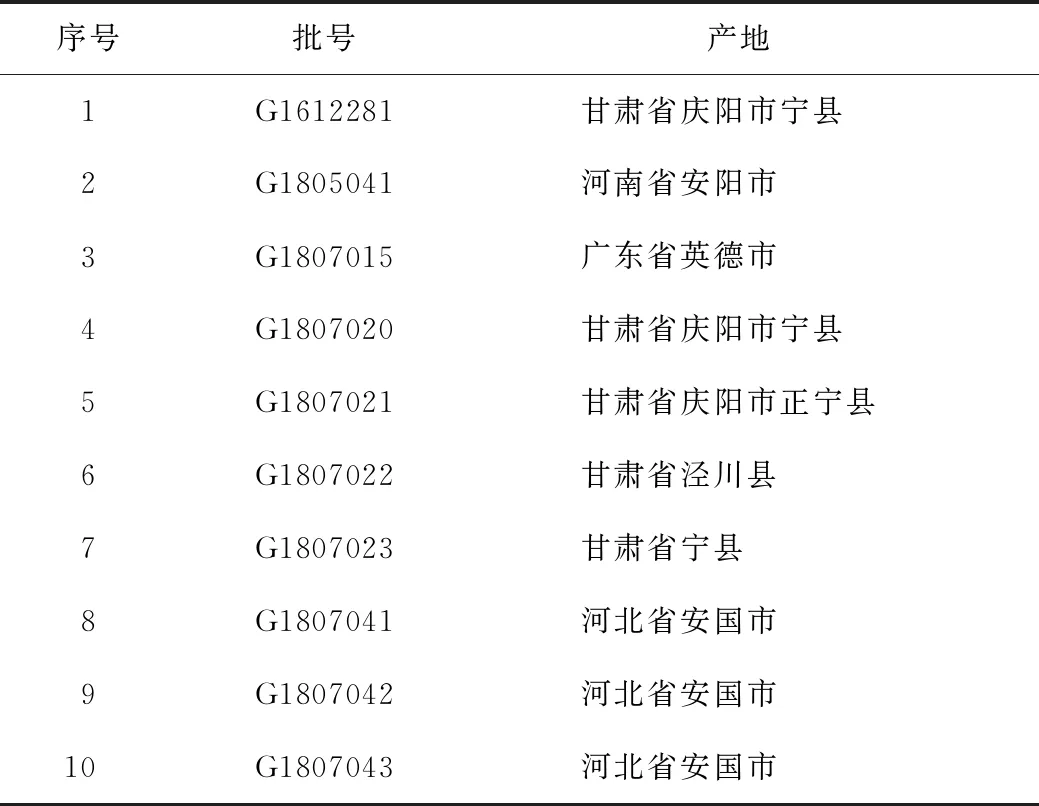
表1 紫苏子样品信息
咖啡酸对照品(批号:110885-201703,纯度:99.7%)、迷迭香酸对照品(批号:110871-201706,纯度:90.5%)、木犀草素对照品(批号:111520-201605,纯度:99.6%)、芹菜素对照品(批号:111901-201603,纯度:99.2%)均购自中国食品药品检定研究院;水为超纯水;甲醇、乙腈为色谱纯;其他试剂为分析纯。
2 方法与结果
2.1 色谱条件
以十八烷基硅烷键合硅胶为填充剂(柱长100 mm,内径2.1 mm,粒径1.8 μm);以乙腈为流动相A,0.1%甲酸溶液为流动相B,按梯度洗脱(0~6 min,10%~18%A;6~15 min,18%~50%A);流速为0.3 mL·min-1;检测波长为330 nm;柱温为30 ℃;进样量为2 μL。
2.2 对照品溶液的制备
精密称取咖啡酸、迷迭香酸、木犀草素及芹菜素适量,加80%甲醇溶液制成含咖啡酸32.662 μg·mL-1、迷迭香酸256.658 μg·mL-1、木犀草素36.364 μg·mL-1、芹菜素37.751 μg·mL-1的混合对照品储备液。再取混合对照品储备液1 mL,加80%甲醇定容于10 mL,得到混合对照品溶液每1 mL含咖啡酸3.266 μg、迷迭香酸25.666 μg、木犀草素3.636 μg、芹菜素3.775 μg。置于4 ℃冰箱中保存,备用。
2.3 供试品溶液的制备
取紫苏子饮片粉末(过二号筛)适量,取约0.5 g,精密称定,置具塞锥形瓶中,精密加入80%甲醇25 mL,称定质量,加热回流60 min,放冷,用80%甲醇补足减失质量,摇匀,滤过,取续滤液,即得。
2.4 方法学考察
2.4.1系统适用性 取上述混合对照品储备液,分别稀释500、1000、4000、8000倍得到混合对照品的定量限(LOQ)及检测限(LOD),见表2,其混合对照品及样品色谱图见图1。各色谱峰分离度良好,经Waters二极管阵列检测器检测,所有目标峰的峰纯度角均小于纯度阈值。表明各色谱峰专属性较好。
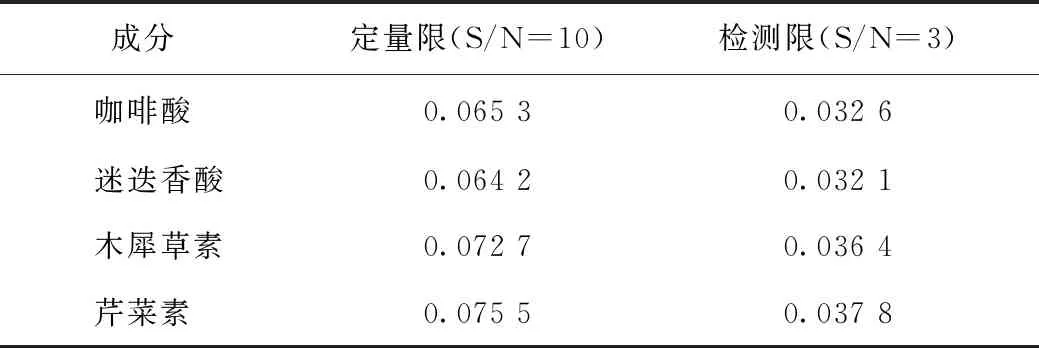
表2 紫苏子中4个成分检测限及定量限结果 μg·mL-1
2.4.2线性考察 分别精密吸取对照品储备液5.0、2.0、1.0、0.5、0.2 mL,加80%甲醇定容于10 mL,摇匀,得到一系列线性对照品溶液,按照2.1项下色谱条件进样,以进样质量浓度X(μg·mL-1)为横坐标,以峰面积Y为纵坐标,绘制标准曲线,计算回归方程,结果见表3。
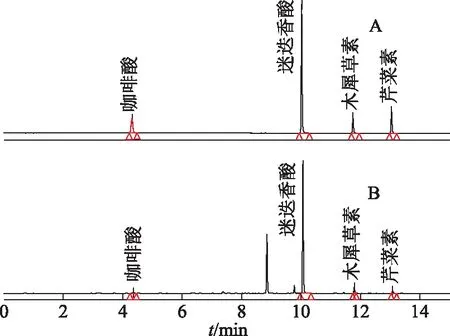
注:A:混合对照品图;B:供试品色谱图。图1 紫苏子混合对照品及样品色谱图
2.4.3精密度考察 精密吸取2.2同一混合对照品溶液,按照2.1项下色谱条件,连续进样6针,计算4种成分峰面积的RSD,结果咖啡酸、迷迭香酸、木犀草素及芹菜素峰面积的RSD(n=6)分别为0.31%、0.28%、0.36%和0.20%。说明仪器精密度良好。
2.4.4稳定性考察 取供试品溶液,按照2.1项下色谱条件,分别于0、2、4、6、8、12 h进样测定,计算4种成分的峰面积的RSD,结果咖啡酸、迷迭香酸、木犀草素及芹菜素峰面积的RSD(n=6)分别为1.37%、0.60%、3.00%和1.70%。说明样品溶液在12 h内基本稳定。
2.4.5重复性考察 称取同一批紫苏子样品(过二号筛)约0.5 g,平行6份,按2.3项下方法制备供试品溶液,按照2.1项下色谱条件分别进样,测定,测得咖啡酸、迷迭香酸、木犀草素和芹菜素的质量分数分别为0.002 9%、0.36%、0.019%、0.006 3%,其RSD(n=6)分别为1.44%、0.91%、3.74%和2.86%,表明方法重复性良好。

表3 紫苏子中4个成分回归方程、相关系数和线性范围
2.4.6加样回收率考察 取同一批已知含量的样品约0.25 g,平行6份,精密称定,置具塞锥形瓶中,按照与所取样品中待测定成分量之比控制在1∶1左右浓度水平精密加入对应的对照品,按2.3项下方法制备供试溶液,按照2.1项下色谱条件进样,测定,计算加样回收率,结果咖啡酸、迷迭香酸、木犀草素和芹菜素的平均加样回收率分别为111.57%、101.97%、95.11%和98.33%,其RSD(n=6)分别为3.32%、0.30%、2.76%和5.40%,说明该方法准确度良好。
2.5 校正因子考察
2.5.1校正因子计算 取2.2项下混合对照品溶液,按照2.1项下色谱条件,分别进样0.2、0.4、0.6、0.8、1.0、2.0 μL,以迷迭香酸为内标,按公式f=As×Ci/(Ai×Cs)(式中Ci为内标物浓度,Ai为内标物峰面积,Cs为组分s浓度,As为组分s峰面积)计算咖啡酸、木犀草素、芹菜素的相对校正因子分别为1.69、1.05、1.42。
2.5.2待测成分的色谱峰定位 色谱峰的准确定位是保证一测多评法应用的前提,本实验以相对保留时间(待测成分与内标成分保留时间之比)作为定位标准,并在不同柱温、不同流速、不同品牌色谱柱及不同色谱仪下对此参数进行考察,结果分别见表4~7,得到不同流速、不同柱温、不同色谱仪条件下咖啡酸、迷迭香酸、木犀草素和芹菜素的相对保留时间波动在3%左右,当色谱柱粒径为1.9 μm时,咖啡酸的相对保留时间偏差较大。因此固定色谱柱粒径为1.8 μm时,结果表明可将此参数用于目标峰的定位。
2.5.3校正因子耐用性考察 由本实验室不同操作人员,在不同实验日期,采用不同实验仪器,取同一份样品,按照供试品溶液制备方法,按照2.1项下色谱条件分别进样,并在不同柱温、不同流速、不同品牌色谱柱及不同色谱仪下对校正因子进行考察,结果分别见表4~7,得到不同流速、不同柱温、不同色谱仪条件下咖啡酸、迷迭香酸、木犀草素和芹菜素的校正因子波动3%左右,当色谱柱粒径为1.9 μm时,校正因子偏差较大。因此固定色谱柱粒径为1.8 μm时,表明方法耐用性良好。
2.6 样品测定
取紫苏子药材10批,按2.3项下方法制备供试溶液,按照分别精密吸取供试品溶液、混合对照品溶液及迷迭香酸对照品溶液各1 μL,按照2.1项下色谱条件进样,测定。通过外标法得到上述4种成分的含量,再按照建立的QAMS进行4个成分色谱峰的定位,并对其含量进行实际测算,计算2种方法所得各成分含量的相对平均偏差(RE),以验证QAMS的合理性。结果见表8。通过2种方法测得的各成分的含量的相对平均偏差<3%,说明该方法的可信度较高。
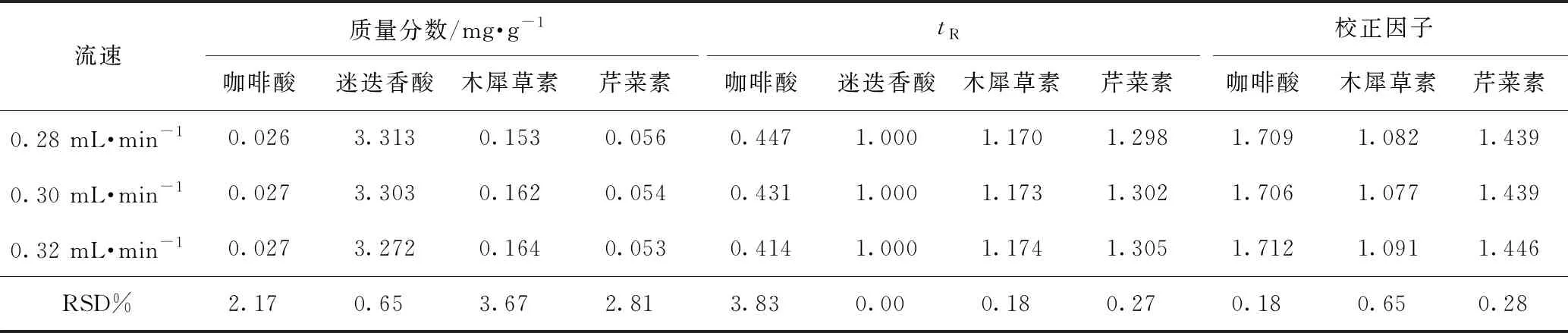
表4 紫苏子中4种成分不同流速的含量测定、相对保留时间及校正因子结果
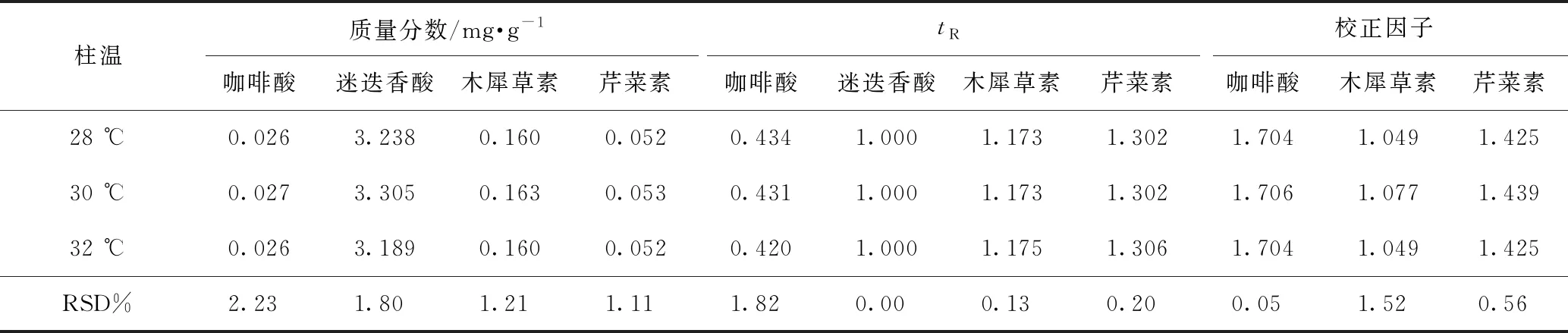
表5 紫苏子中4种成分不同柱温的含量测定、相对保留时间及校正因子结果
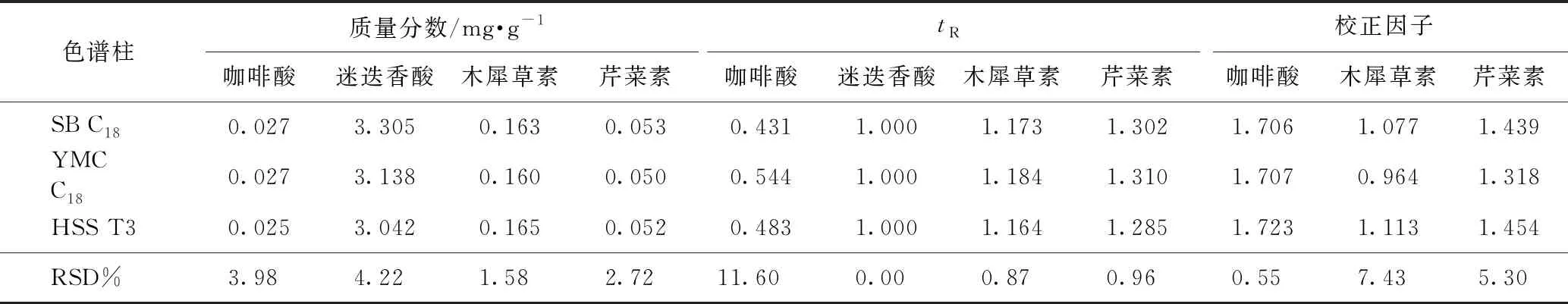
表6 4种成分不同色谱柱的含量测定、相对保留时间及校正因子结果
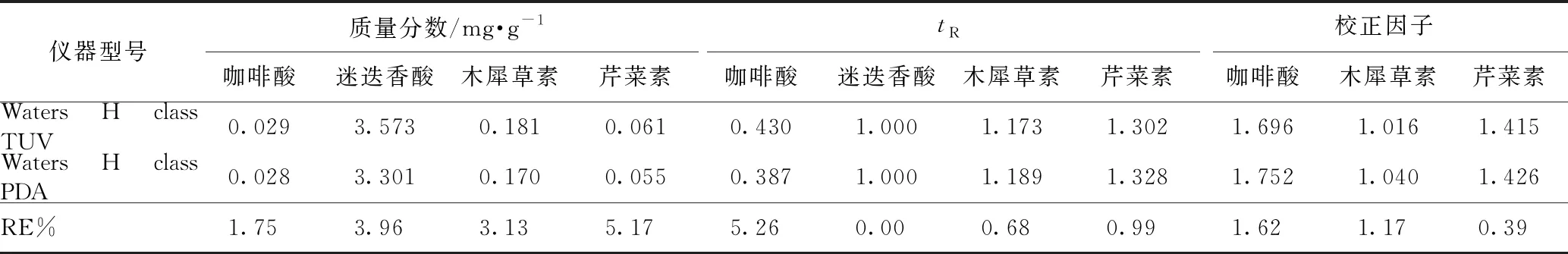
表7 4种成分不同色谱仪的含量测定、相对保留时间及校正因子结果
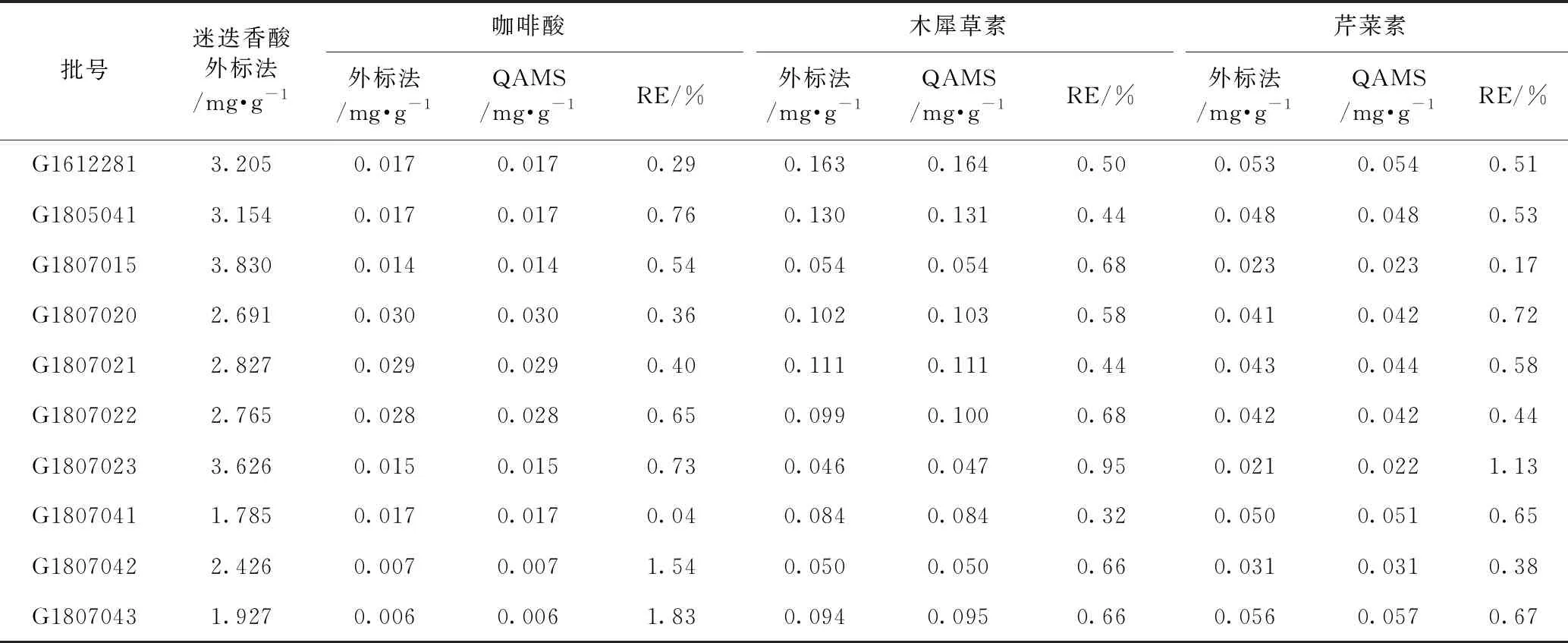
表8 不同产地紫苏子中4种成分样品测定结果(外标法和QAMS)
2.7 不同产地紫苏子药材的聚类分析
基于QAMS的测定结果,对上述10批不同产地的紫苏子药材进行分析,通过SPSS V20版软件进行系统聚类,得到聚类分析结果见图2。
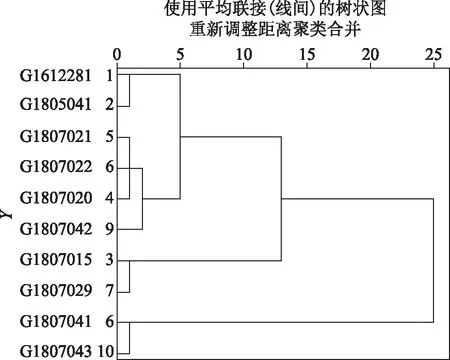
图2 紫苏子药材聚类分析结果
从上图可知,以欧氏距离<10,可将10批紫苏子药材分为3类,其中G1612281、G1805041、G1807021、G1807022、G1807020、G1807042为第一类,G1807015、G1807023为第二类,G1807041、G1807043为第三类,第一类的产地分布较多,包括河南省安阳市龙安区、甘肃省庆阳市宁县、甘肃省庆阳市正宁县、甘肃省泾川县、河北省安国市,第二类产地包括广东省英德市大湾镇、甘肃省庆阳市宁县,第三类河北省安国市,主要为不存在明显的产地差异性,进一步分析各类之间的含量配比,以迷迭香酸-木犀草素-芹菜素三者含量比例关系来看,第一类的迷迭香酸-木犀草素-芹菜素接近66∶2.5∶1,第二类的接近165∶2∶1,第三类的接近34∶1.6∶1,由此来看,紫苏子药材不同各产地成分间的差异可能较小,提示以不同成分的含量配比之间可能与其植物变异及种源鉴定间存在一定关联。
3 讨论
《中华人民共和国药典》2015年版紫苏子项下药材含量测定以迷迭香酸为指标,从各成分含量测定的结果来看,其含量较高,且易于得到,因此,选迷迭香酸作为内标,基于咖啡酸、迷迭香酸、木犀草素、芹菜素均值330 nm处有最大吸收,因此建立上述2种酚酸类及黄酮类成分的含量测定QAMS。本研究分别考察了不同料液比、不同提取溶剂、不同提取方式及不同提取时间对4种成分的含量,得到当料液比为0.5∶25 g·mL-1,以80%甲醇作为提取溶剂,加热回流处理60 min时,所得提取效率较高。
唇形科紫苏属1年生草本植物苏Perillafrutescens(L.) Britt.有1个种、3个变种和7个变型,紫苏有叶两面紫色(其种子简称为双紫苏子)和面绿背紫(其种子简称为单紫苏子)2个变型,有文献报道两者之间含量差异较大[7]。但以种子入药后,因缺乏对原植物的形态鉴定,单一的含量测定指标,难以对种子的变异或者对种子规格进行区分。本研究通过建立一测多评的方法,得到10批紫苏子的咖啡酸质量分数为0.000 6%~0.003%,迷迭香质量分数为0.18%~0.38%,木犀草素质量分数为0.005%~0.016%,芹菜素质量分数为0.002%~0.006%,并对不同成分之间的含量比例关系进行分析,为种源研究提供了新思路。
猜你喜欢
北京联合大学学报(2022年1期)2022-02-12
中国外汇(2019年10期)2019-08-27
中国外汇(2019年10期)2019-08-27
中国外汇(2019年22期)2019-05-21
中国外汇(2019年21期)2019-05-21
分析化学(2018年12期)2018-01-22
西江月(2017年4期)2017-11-22
红豆(2017年1期)2017-01-14
小雪花·成长指南(2016年10期)2016-11-01
中国民族民间医药·下半月(2014年5期)2014-12-02
