超高效液相色谱-四极杆-飞行时间高分辨质谱用于乳液类化妆品中9种抗过敏违禁药物的筛查和定量分析
2020-11-02 01:39王梦颖陈烨超涂凤琴卢跃鹏王煜红
色谱 2020年12期
王梦颖, 陈烨超, 涂凤琴, 侯 靖, 杨 明, 卢跃鹏, 王煜红, 杨 总, 陈 丹*
(1. 武汉食品化妆品检验所, 湖北 武汉 430014; 2. SCIEX亚太技术支持中心, 上海 200050)
随着人们对生活品质的追求日益提高,具有清洁、护肤、美容和修饰等功能[1]的化妆品成了人们日常生活必需品,但是彩妆及清洁类护肤品的过度使用容易让皮肤变得敏感,受损皮肤在雾霾、花粉或恶劣气候的持续暴露下容易出现红肿、脱皮、发痒等不良反应。不法商家为了抓住敏感肌肤人群的消费市场,在化妆品中非法添加一些抗组胺类抗过敏药物,并宣称其商品具有镇静舒缓、修复敏感等功效,但抗组胺类药物在临床上主要用于荨麻疹、湿疹、过敏性皮炎等皮肤病以及过敏性鼻炎、过敏性哮喘等呼吸道疾病,含有抗过敏药物化妆品的长期使用容易造成药物依赖性皮炎、皮肤色素沉积、角质层变薄及皮炎等问题[2],商家在化妆品中非法添加抗过敏药物的行为严重损害了消费者利益。2015年11月国家食品药品监督管理总局颁布的《化妆品安全技术规范》中明确规定9种抗组胺类抗过敏药物(异丙嗪、羟嗪、奋乃静、西替利嗪、氟奋乃静、氯丙嗪、氯雷他定、特非那定、赛庚啶)不得作为生产原料和组分添加到化妆品中[3]。为了保证化妆品质量安全,保障消费者身体健康,需要加强对化妆品中抗过敏物质的筛查分析。
目前抗组胺类抗过敏物质的常用检测方法主要包括液相色谱(HPLC)法[4,5]、气相色谱(GC)法[6]、液相色谱-串联质谱(LC-MS/MS)法[2,7]等。LC-MS由于具有快速简便、选择性好等特点被广泛用于非法添加检验领域,但其也存在分辨率较低,易出现假阳性等不足。而超高效液相色谱-四极杆-飞行时间高分辨质谱(UPLC-Q-TOF-HRMS)技术具有高质量精度、全质量数据采集和数据库检索等优势[8],其高分辨率能提高定性筛查能力,全质量数据采集模式能降低阳性样品遗漏,基于化合物二级碎片图谱信息的数据库检索功能能够增加定性准确率,同时信息非依赖性采集(SWATH, sequential window acquisition of all theoretical mass spectra)与二级子离子定量技术相结合可实现化合物的准确定量。随着UPLC-Q-TOF-HRMS技术的日益成熟,其精确相对分子质量定性和定量技术的优势能为化妆品中违禁使用药物的测定提供有效解决方案。
本实验采用固相萃取净化技术结合高分辨质谱检测,建立了基于UPLC-Q-TOF-HRMS技术的乳液类化妆品中9种抗过敏药物的筛查和定量方法。
1 实验部分
1.1 仪器、试剂及材料
X-500R超高效液相色谱-四极杆-飞行时间质谱仪(美国SCIEX公司), ME204分析天平(瑞士Mettler toledo公司), Vortex-Genie2涡旋振荡仪(美国Scientific Industries公司), Allegra 64R高速冷冻离心机(美国Beckman coulter公司), Milli-Q超纯水机(德国Merck Millipore公司),固相萃取装置(美国安捷伦公司),超声波清洗机(英国Prima公司)。
包含盐酸异丙嗪、盐酸羟嗪、奋乃静、盐酸西替利嗪、氟奋乃静、盐酸氯丙嗪、氯雷他定、特非那定、赛庚啶等9种抗过敏物质的混合液体标准品,每种化合物质量浓度均在100 μg/mL左右(天津阿尔塔科技), Oasis PRiME HLB固相萃取小柱(200 mg, 3 mL,美国Waters公司), LC-C18固相萃取小柱(200 mg, 3 mL,德国CNW Technologies公司), Oasis HLB固相萃取小柱(200 mg, 6 mL,美国Waters公司),甲醇、乙腈和甲酸为色谱纯(德国Merck公司),实验用水均为经Milli-Q超纯水机纯化后的高纯水。
1.2 标准溶液配制
准确量取质量浓度均约为100 μg/mL的混合标准品母液1 mL于100 mL容量瓶中,用乙腈定容至刻度线,得到质量浓度为1.00 μg/mL的混合标准使用液A,取A溶液1 mL于10 mL容量瓶中,用乙腈定容至刻度线,得到质量浓度为0.100 mg/L的混合标准使用液B。
分别准确移取B混合标准溶液0、10、20、50、100 μL, A混合标准溶液20、50、100、200 μL于进样小瓶中,用乙腈溶液定容至1 mL,得到质量浓度分别为0、1、2、5、10、20、50、100、200 μg/L的纯溶剂标准工作溶液。
1.3 样品前处理
准确称取0.50 g乳液样品于10 mL离心管中,加入4 mL乙腈,涡旋1 min,超声处理10 min后于冷冻离心机中10 000 r/min离心10 min,取上清液过PRiME HLB柱,将流出液过0.22 μm有机滤膜后上机检测。
1.4 仪器条件
1.4.1色谱条件
色谱柱:Waters XBridge C18(150 mm×2.1 mm, 5 μm);柱温:40 ℃;进样量:10 μL;流动相:A相为0.1%甲酸水溶液,B相为乙腈;梯度洗脱程序:0~1.0 min, 3%B; 1.0~1.5 min, 3%B~15%B; 1.5~7.0 min, 15%B~45%B; 7.0~19.0 min, 45%B~98%B; 19.0~22.0 min, 98%B; 22.0~22.1 min, 98%B~3%B; 22.1~25.0 min, 3%B;流速:0.5 mL/min。
1.4.2质谱条件
离子源:电喷雾离子源;扫描模式:正离子扫描;母离子扫描范围:m/z100~1 000;离子源温度:550 ℃;喷雾电压:5 500 V;去簇电压:65 V;雾化气:379.22 kPa (55 psi);辅助雾化气:379.22 kPa (55 psi);气帘气:241.32 kPa (35 psi)。质谱数据采集模式:采用信息依赖性采集模式(IDA)进行标准谱库信息采集,利用SWATH模式进行样品质谱数据采集。采用SCIEX OS 1.5软件(美国SCIEX)进行数据采集和处理。
1.5 化合物标准谱库建立和定性、定量分析方法
准备质量浓度为50 μg/L包含9种目标化合物的混合标准溶液进行上机检测,采用IDA采集数据,得到9种化合物的精确相对分子质量、保留时间、同位素峰、特征碎片离子质谱图,建立包含上述信息的化合物标准谱库,9种化合物的标准信息见表1。
实验样品经前处理后进样检测,将采集得到的数据导入SCIEX OS中数据分析模块,将样品质谱信息与自建的化合物标准谱库进行比对。在相同的实验条件下,如果被测样品在提取相应分子离子的质量色谱图中出现了与标准品保留时间一致的色谱峰、精确质量数偏差不高于5×10-6(5 ppm)、同位素丰度比差异不高于10%、二级质谱图谱库匹配得分大于70分且两个定性子离子丰度比误差低于20%时,则可判断被测样品中存在相应的化合物[9]。上述定性判定依据满足欧盟法规(SANTE/11945/2015)中关于高分辨质谱应用于定性分析时的判定,即每个化合物对应两个质量精度在5×10-6以内的离子且两个离子的丰度比误差低于30%的规定。将样品中被测化合物定量离子提取色谱峰的峰面积代入相应的标准曲线中,计算得到样品中被测化合物的浓度。

表 1 9种抗过敏药物的Q-TOF-HRMS数据库
1.6 基质效应评价
基质效应在质谱分析中普遍存在,特别是在电喷雾电离模式下,共流出干扰物会对目标离子造成不可预期的离子抑制或增强效应[10]。本实验中通过基质匹配标准曲线与溶剂标准曲线的参数对比来评价护肤水的基质效应。准确称取0.50 g空白乳液样品于10 mL离心管中,按1.3节中方法处理样品,得到空白基质溶液。取7支小瓶,分别加入0、5、10、20、50、100、200 μL混合标准使用液A,用空白基质溶液定容至1 mL,得到质量浓度分别为0、5、10、20、50、100、200 μg/L的基质标准溶液,将上述基质标准溶液与1.2节中溶剂标准溶液一起进行上机检测,得到空白基质和纯溶剂标准溶液的标准曲线。基质效应η=(基质匹配曲线斜率-溶剂曲线斜率)/溶剂曲线斜率×100%[11,12],η的绝对值随着基质效应的增强而变大,η为正值说明基质增强,负值则为基质抑制。当η绝对值<20%时表示基质效应微弱,η绝对值在20%~50%范围内时表示具有中等强度基质效应,基质效应绝对值大于50%时表示基质效应较强[13,14]。
2 结果与讨论
2.1 仪器方法优化
2.1.1色谱方法
对9种化合物进行初步的正负离子扫描发现,在ESI+模式下可得到[M+H]+的分子离子峰,因此选择正离子扫描模式,并尝试在流动相中加入一定量的甲酸以增强离子化效率。实验比较水-乙腈与0.1%甲酸水-乙腈作为流动相体系时化合物的分离情况和响应强度,结果见表2,表中数据显示在加入甲酸的情况下大部分化合物母离子峰响应提高,因此选择0.1%甲酸水-乙腈作为流动相,并根据化合物出峰情况调整流动相洗脱梯度。结果表明,在1.4节方法下9种化合物均能出峰,且峰形良好。
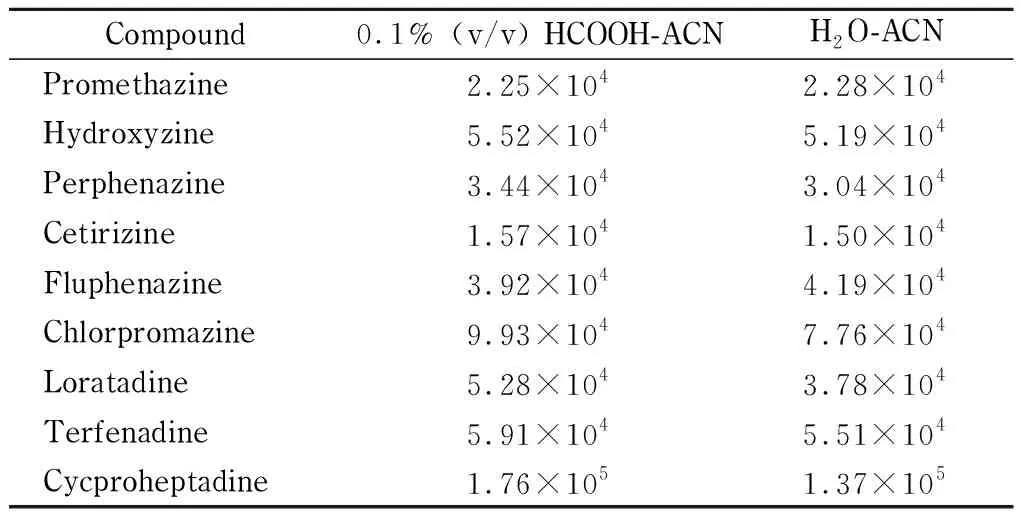
表 2 两种流动相体系下9种化合物的提取离子响应强度
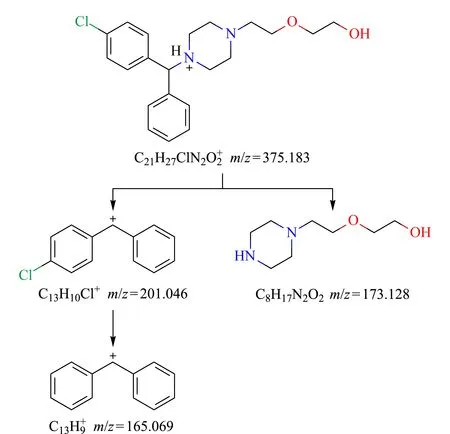
图 1 羟嗪的二级碎片裂解途径Fig. 1 Fragmentation pathway of hydroxyzine

图 2 不同体积乙腈和甲醇作为提取溶剂时加标样中9种化合物的响应强度Fig. 2 Response intensities of the nine compounds in spiked samples extracted with different volumes of acetonitrile and methanol
2.1.2质谱方法
为了提高数据采集效率,实验中采用信息非依赖性SWATH扫描模式。与达到判定条件才能触发二级扫描的信息依赖性IDA模式不同,它是一种全景式、连续性、高通量的数据非依赖性质谱数据获取方式,它通过对化合物母离子的质量范围进行智能窗口划分,采集每个窗口内所有母离子的二级碎片,然后通过软件去卷积技术进行二级碎片归属。在本实验中首先用SWATH模式采集一个空白基质样品,得到样品的总离子流信息,对在1~25 min内检测到的所有化合物依照质量数的大小进行排序,通过软件根据相对分子质量对其进行连续平均分段,以该分段为依据设置SWATH扫描参数[15,16]。样品中的化合物在色谱分离、质谱检测后得到详细化合物分子信息,根据母离子峰保留时间、精确质量数和同位素峰比值可以确定元素组成,由二级子离子碎片图谱可以判定化合物的分子组成和裂解途径,图1展示了羟嗪的裂解途径。
2.2 前处理方法优化
2.2.1提取溶剂选择
实验中比较了不同体积乙腈和甲醇对乳液类样品中目标化合物的提取效果。样品中分别加入等量乙腈和甲醇,涡旋静置后发现,乙腈提取组中离心管底部出现明显颗粒沉淀,分析出现这种情况的原因可能是由于乳液主要由水、甘油等多元醇、低碳脂肪酸、具有保湿功效的低聚糖、氨基酸、植物提取液等成分组成[17],乙腈能改变其中的部分低聚糖、多糖和氨基酸的溶解性,使其沉淀析出,这在一定程度上有助于乳液类基质中杂质的去除。随后考察了4~8 mL的乙腈和甲醇对提取效果的影响,实验结果见图2,可以看出,随着提取体积的增加,9种目标物的响应强度均有相似的下降趋势,且整体而言乙腈的提取效果优于甲醇,综合经济成本和提取效率考虑,选择4 mL乙腈作为提取溶剂。
2.2.2净化条件优化
样品经乙腈提取后,部分盐及脂质会随化合物一并被提取出来,为减少它们对检测结果的影响,实验考虑采用固相萃取小柱对提取液进行净化,期望提取液通过净化柱后,脂质等杂质被保留在柱上,目标物随提取液流出,随后便可将提取液直接过膜上机,免去洗脱、氮吹置换溶剂等复杂操作,以达到简单快速净化的目的。于是考察了C18、HLB及PRiME HLB这3种小柱的净化效果,结果经C18和HLB固相萃取小柱净化后的提取液中奋乃静和氯丙嗪没有回收,而经PRiME HLB处理后,提取液中目标化合物均能出峰,由表3中数据可以看出,经PRiME HLB净化后,9种目标物均具有较高的响应强度。同时采用PRiME HLB固相萃取柱对提取液进行净化,能在一定程度上降低杂质对目标色谱峰的干扰。以异丙嗪为例,从图3可以看到,与未净化的提取液相比,经PRiME HLB净化后的提取液中化合物附近干扰峰减少,所以确定前处理采用4 mL乙腈作为提取溶剂,以PRiME HLB固相萃取柱进行提取液净化。
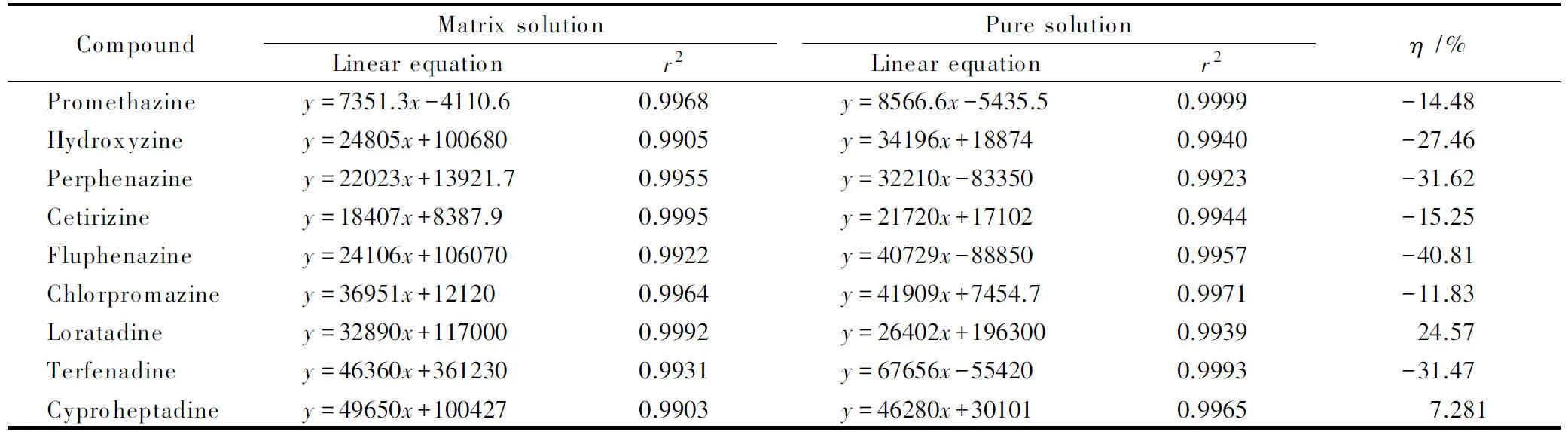
表 4 9种抗过敏药物采用一级母离子定量时的标准曲线和基质效应
采用UPLC-Q-TOF-HRMS以SWATH方式进行数据采集后,可以采用母离子峰面积定量或二级子离子峰面积定量,前者计算方便,是常规的定量方式,但存在抗基质效应能力弱,易被临近峰干扰的风险。后者是基于SWATH连续数据采集模式下的定量方式,采用高精度二级碎片定量分析选择性更强。本实验中采用上述两种定量方式分别对空白基质溶液和纯溶剂配制标准曲线进行定量分析,观察曲线线性关系并比较在不同定量方式下基质效应的强弱,结果见表4及表5。
由表中数据可知,9种化合物在不同基质中采用两种方式定量时均具有良好线性,线性相关系数大于0.99,线性范围为5~100 μg/L。当采用母离子定量时,基质对9种化合物均有一定程度的影响,其中对氟奋乃静的基质抑制效应达到了40.81%。而采用子离子碎片定量后,基质效应减弱,η绝对值控制在20%以内。为了增加定量准确度,本方法选择采用纯溶剂配制标准曲线结合子离子峰面积定量进行样品中目标物质浓度分析。

表 3 加标样品经PRiME HLB净化前后提取母离子色谱峰的响应强度
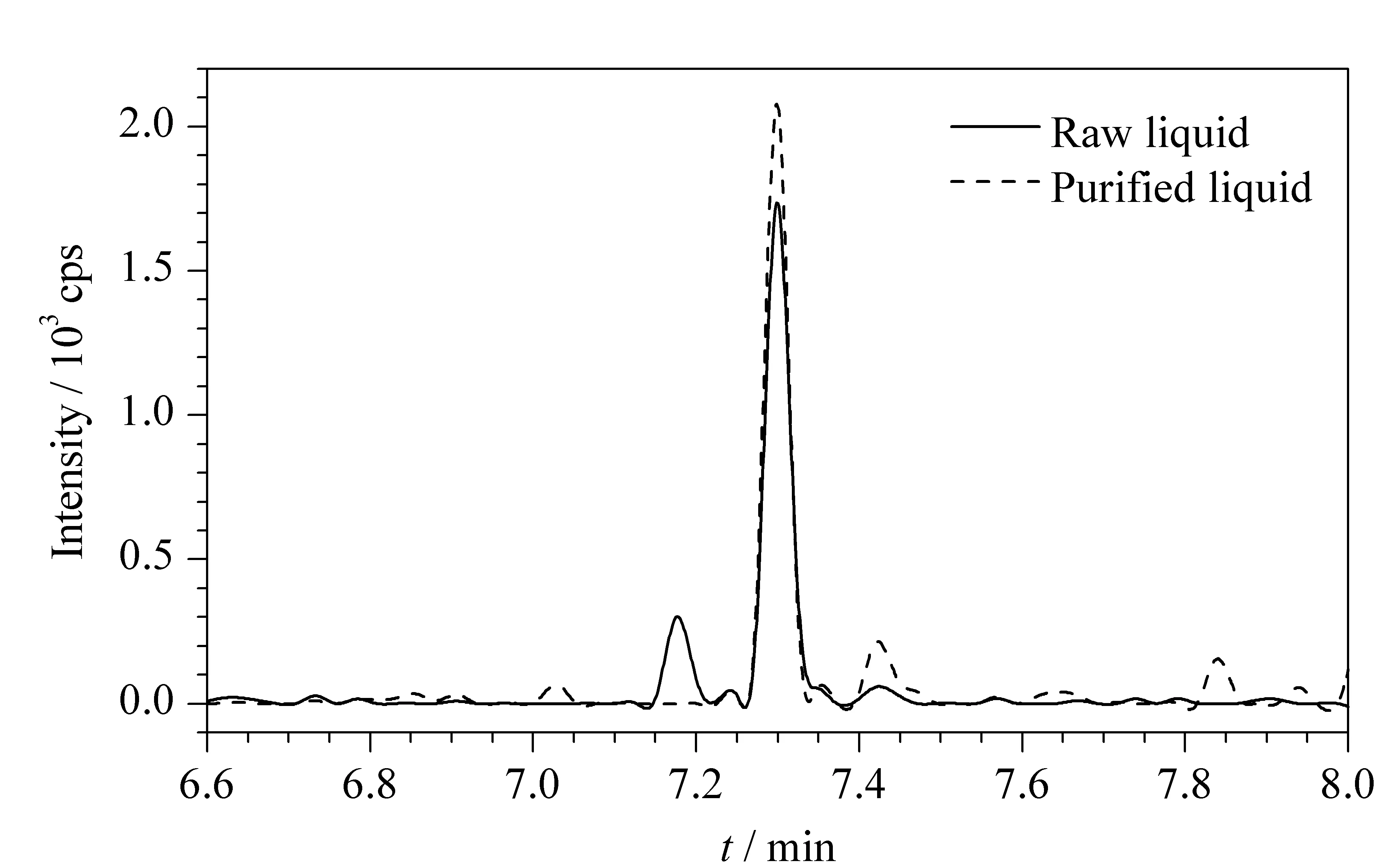
图 3 加标样品经PRiME HLB净化前后异丙嗪的提取母离子色谱峰图Fig. 3 Extracted ion chromatograms of promethazine in spiked sample before and after PRiME HLB processing
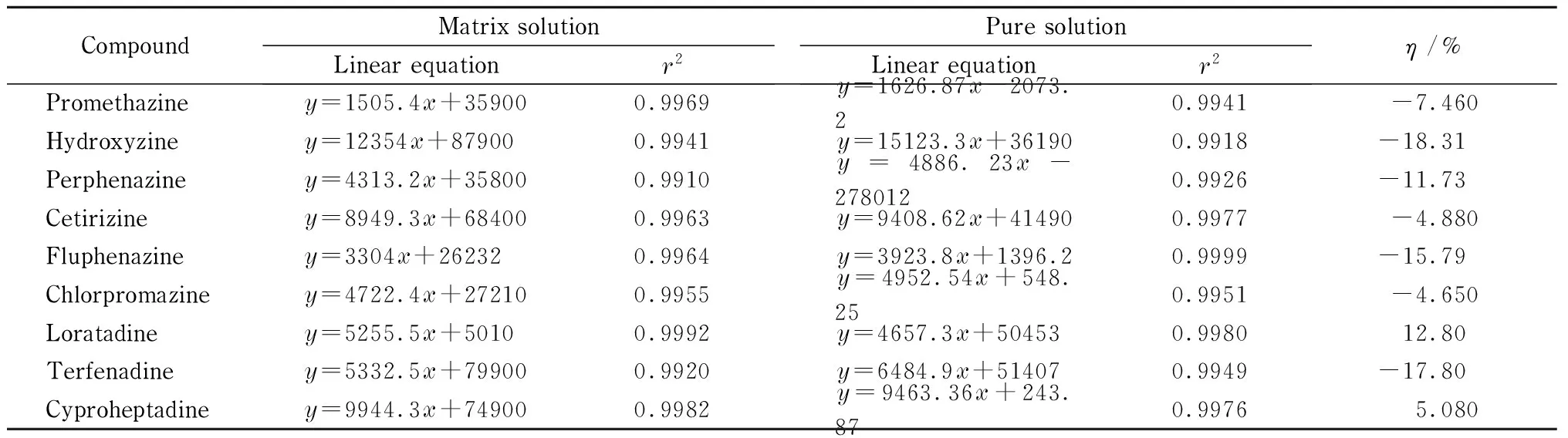
表 5 9种抗过敏药物采用二级子离子定量时的标准曲线和基质效应
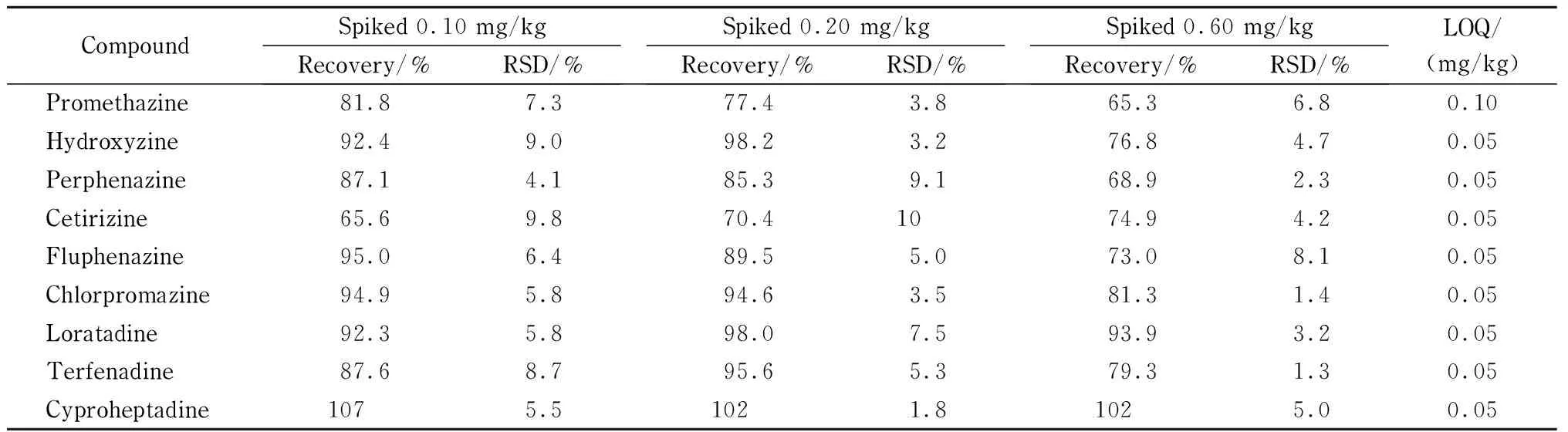
表 6 9种抗过敏物质在乳液类基质中的加标回收率、RSD和LOQ (n=6)
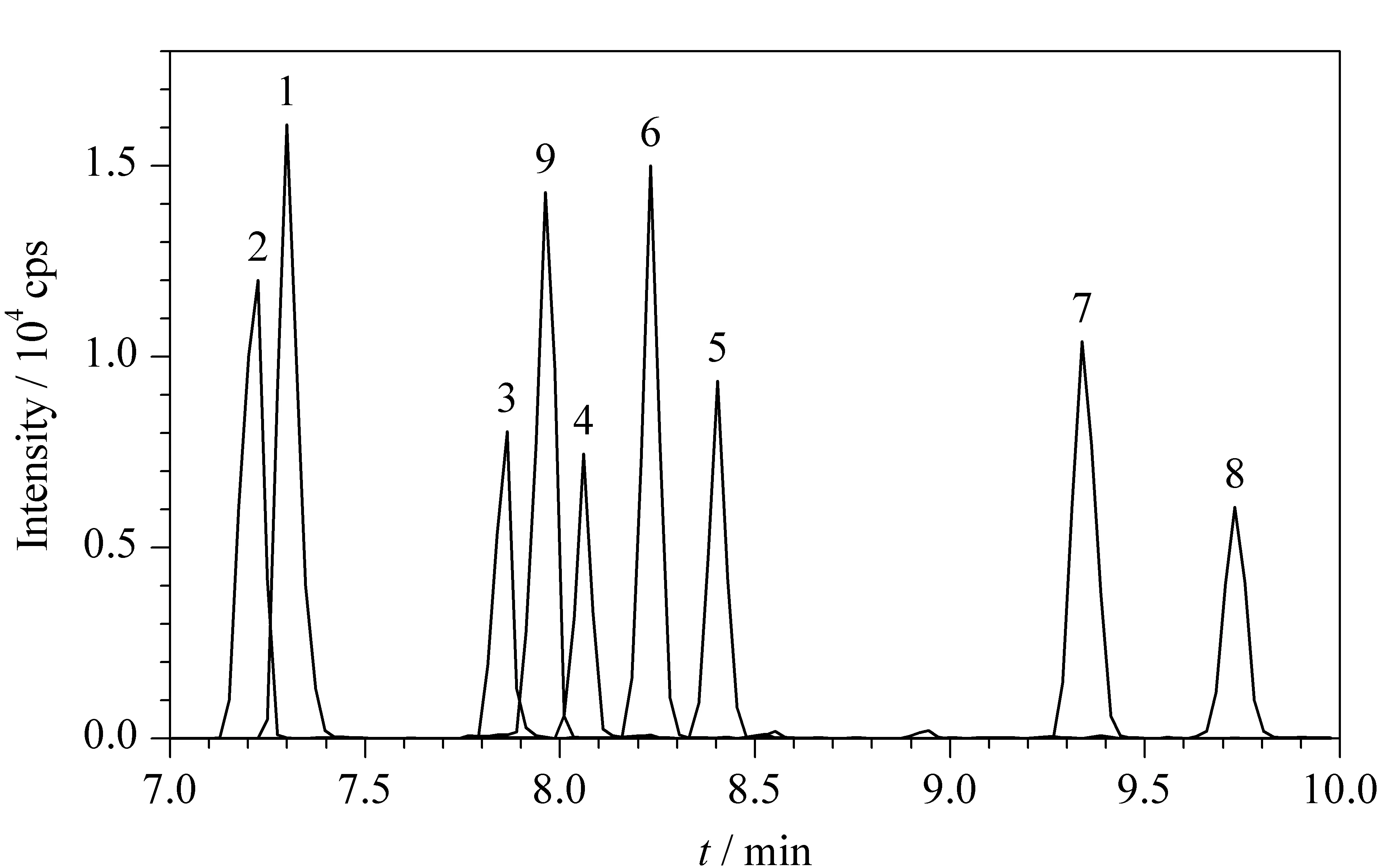
图 4 加标样品中9种抗过敏化合物的提取离子色谱图Fig. 4 Extracted ion chromatograms of the nine antiallergic compounds in spiked sample Peak identifications: 1. promethazine; 2. hydroxyzine; 3. perphenazine; 4. cetirizine; 5. fluphenazine; 6. chlorpromazine; 7. loratadine; 8. terfenadine; 9. cyproheptadine.
在0.50 g空白乳液样品中分别添加50、100、300 ng混合标准物质,制备加标量分别为0.10 mg/kg、0.20 mg/kg和0.60 mg/kg的加标样品,每组进行6次平行实验,加标量为0.20 mg/kg样品中9种目标化合物的提取母离子色谱图见图4,加标回收定量结果见表6。由图4可以看出,9种目标物均能够正常出峰,响应强度高,且具有较好的分离度。由表6中数据可以看到,当添加水平为0.10 mg/kg时,9种化合物的加标回收率为65.6%~107%,相对标准偏差(RSD)为4.1%~9.8%;在添加水平为0.20 mg/kg时,加标回收率为70.4%~102%, RSD为1.8%~10%;在添加水平为0.60 mg/kg时,加标回收率为65.3%~102%, RSD为1.3%~8.1%,在3个不同水平下加标回收率均能满足实验日常检测的要求。
采用标准添加法来确定方法定量限,以10倍信噪比下各待测物质的质谱响应值来计算乳液基质中9种抗过敏物质的定量限,结果见表6, 9种抗过敏物质中除异丙嗪的定量限为0.10 mg/kg以外,其他8种物质的定量限均为0.05 mg/kg,低于《化妆品安全技术规范2015版》[3]中基于LC-MS/MS的抗过敏物质测定方法中的定量限(0.50 mg/kg),本方法具有更高的灵敏度。
2.5 实际样品测定
采用本方法对市售的32份乳液样品中9种抗过敏物质进行了测定,结果未出现阳性样品,分析原因可能是此次检测的样品均为知名品牌产品,产品质量有一定的保障。后期将对不同层次品牌进行检测,扩大抽检样本量。
3 结论
本文建立了基于UPLC-Q-TOF-HRMS的乳液类化妆品中9种抗过敏物质的筛查和定量分析方法,采用基于PRiME HLB小柱的“一步式”净化方法,提取液经过小柱后,不需要进行后续淋洗和洗脱,可直接过膜上机,与王燕芹等[2]采用的PCX小柱净化法相比,该处理更简便,耗时更短。采用SWATH数据采集模式结合碎片离子峰面积定量,在降低基质效应的同时保证了定性定量结果的准确度。该方法灵敏度、准确度、精密度和线性关系良好,操作简便快捷、灵敏可靠,可用于乳液类化妆品中违禁添加抗过敏物质的快速筛查和准确定量。
猜你喜欢
煤化工(2022年3期)2022-07-08
食品安全导刊(2021年20期)2021-08-30
色谱(2021年7期)2021-06-07
中国粮油学报(2019年4期)2019-07-12
中成药(2018年2期)2018-05-09
天然产物研究与开发(2018年1期)2018-02-02
中成药(2017年4期)2017-05-17
山东工业技术(2016年10期)2016-09-06
中国资源综合利用(2016年10期)2016-01-22
特产研究(2014年4期)2014-04-10