
DNA合成、组装与纠错技术研究进展
2021-01-21 01:55彭凯逯晓云程健刘莹江会锋郭晓贤
合成生物学 2020年6期
彭凯,逯晓云,程健,刘莹,江会锋,郭晓贤
(1 中国科学院天津工业生物技术研究所系统微生物工程重点实验室,天津 300308;2 天津科技大学生物工程学院,天津 300457)

合成生物学是继DNA 双螺旋发现催生分子生物学,“人类基因组计划”实施催生基因组学后的第三次生物技术革命。DNA 合成技术是合成生物学的核心使能技术之一。随着我们对生命系统认识的深入,生命体系的重新设计和创造已成为生物学领域最富想象力和活力的研究领域。大规模基因组DNA 设计和合成赋予我们改造细胞功能甚至创造人工生命的能力,有助于提高我们对生命体的理解、预测和操控的能力[1-5]。
自20 世纪50 年代以来,大量科研工作者尝试通过化学和酶促方法合成DNA。首先获得成功的是化学合成技术。经过多年优化和改进,DNA 化学合成经历了从柱式合成到芯片合成的变革发展,并得到了广泛的市场化应用。但是现有化学方法的偶联效率和副反应使寡核苷酸合成长度局限于200~300 nt,难以到达 kb 级的基因长度[6-8]。因此更长片段则需通过组装技术拼接寡核苷酸片段,直至获得基因、染色体或基因组长度的DNA[9]。然而,寡核苷酸片段合成和DNA 组装过程会产生很多错误,降低长片段DNA 的正确率。纠错技术的应用可去除DNA 合成和组装过程引入的大量错误,进而降低正确DNA 片段的筛选与测序成本[10]。本文作者将重点综述DNA 合成、组装与纠错技术相关的研究进展,以此期望促进我国DNA合成相关技术创新发展。
1 DNA合成技术
1.1 寡核苷酸柱式合成技术
寡核苷酸的化学合成始于20 世纪50 年代,于80 年代开发出亚磷酰胺三酯化学合成法[11],并应用于柱式合成,在90 年代又应用到基于芯片的高通量合成技术中[12]。亚磷酰胺三酯合成法由脱保护(deprotection)、偶联(coupling)、加帽(capping)和氧化(oxidation)四步化学反应组成循环,通过分步活化结合在核苷酸3'位和5'位的化学活性保护基团实现可控合成,在固相载体上从3'到5'方向逐个延伸合成寡核苷酸链[13](图1)。
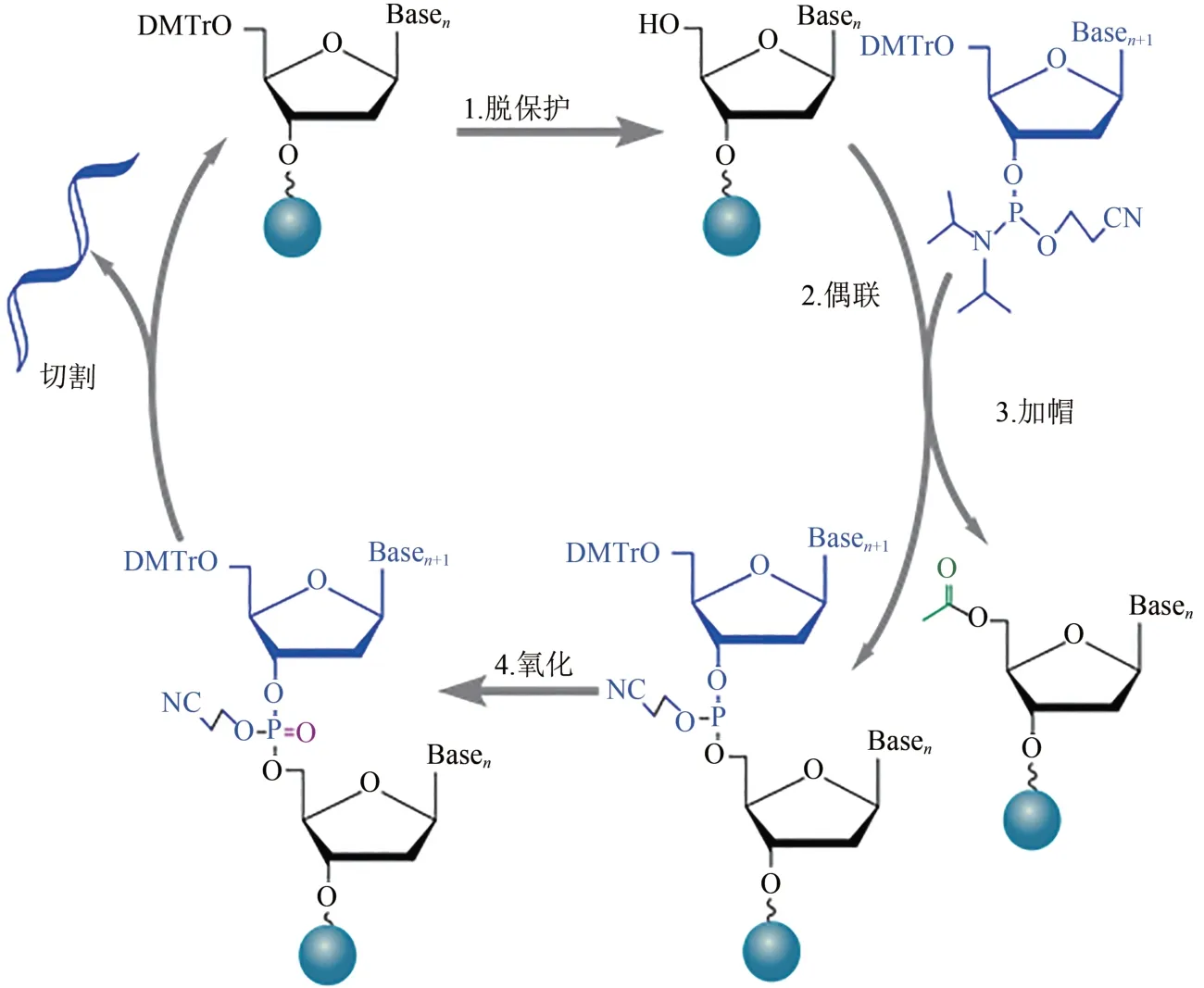
图1 固相亚磷酰胺寡核苷酸合成反应循环Fig.1 Reaction cycle for solid-phase phosphoramidite synthesis of oligonucleotide
基于亚磷酰胺三酯合成法的柱式DNA 合成技术,以填充多孔玻璃(controlled pore glass,CPG)或聚苯乙烯(polystyrene,PS)筛板的合成柱作为固相载体,带保护的单体经过化学试剂分步活化被逐个按序添加到固定在合成柱的引发剂上。目前商品化的柱式DNA合成反应偶联效率可达98%~99.8%,错误率约为1/600 nt,产量一般在1 µmol以内,单个循环耗时6~8 min。权衡效率与成本后最长合成长度通常控制在100 nt 左右[14]。若将合成柱固定到多孔装载板上,单轮合成通量可提高到1536 根,平均偶联效率为99.5%,错误率约1.53/717 nt,成本大约 0.277美分/碱基[15]。
柱式DNA 合成技术发展至今,自动化合成设备成熟。能够方便、灵活地提取用于合成某段基因所需的任意寡核苷酸片段,满足一般实验的要求。随着合成生物学领域对大规模基因和基因组合成需求的日益高涨,柱式合成持续合成能力弱、化学试剂耗费大、副反应多、通量低等不足也日益凸显[14]。为突破低效率、低通量、高成本的限制,寻求DNA 合成技术持续发展的研究主要致力于:①开发具备高反应通量、多重功能的集成芯片作为固相载体,并行合成寡核苷酸;②开发基于无模板单链DNA合成的酶促寡核苷酸合成技术。
1.2 芯片DNA合成技术
芯片可作为DNA 合成固相载体,以高密度、集成方式在其表面特定位点上进行合成反应,从而在节省试剂的同时实现高通量合成。芯片DNA合成技术仍以亚磷酰胺合成法四步反应循环为基础,但采用不同的“脱保护”定点控制方法,分别发展出了光刻合成、电化学合成和喷墨打印合成等DNA合成平台[12]。
最早出现的光刻合成通过精确控制光在芯片表面指定位点的投射,分解光敏保护基团或光敏催化剂产酸进行脱保护,实现核苷酸在不同合成位点的有序添加。根据光照控制方式又分为掩模光刻合成[16-17](为Affymetrix 采用)与无掩模光刻合成[18-20](为 NimbleGen 和 LC Sciences 采用)。电化学合成利用电化学反应产生酸,控制聚合物膜上常规亚磷酰胺单体的加成[21],被CustomArray(已被Genscript收购)所采用。Agilent的喷墨打印合成则通过将酸溶液或试剂喷射在反应位点催化脱保护[22-23]。
芯片DNA 合成的通量高,通过精密的自动化控制,芯片上单个微反应室能以皮升级的反应体系进行合成反应。不同芯片合成通量在3×103~3×106之间,合成密度在 105~106/cm2,成本低至0.001~0.1 美分/碱基。与柱式合成相比,在进行大规模基因合成时,芯片合成法占有绝对优势。但芯片制作工艺复杂,随着合成密度的增加,对芯片生产技术以及合成仪的自动化控制技术要求也高。此外,芯片合成还存在由定点错位或试剂隔离不良而产生“边缘效应”、脱保护反应不彻底、脱嘌呤等副反应多的问题[6]。这些因素直接影响合成序列的完整性和正确性,使DNA 芯片合成的效率在90%~99%,合成长度限制在25~200 nt。芯片单个微量反应体系合成的寡核苷酸产量低(约为10-15mol),错误率也高于柱式合成,难以单独分离纯化。因此,不适用于单基因或小规模的基因、常规探针以及引物合成。
1.3 酶促DNA合成技术
DNA 化学合成中大量使用有毒、易燃且不稳定的有机试剂,环境友好度低,因而生物合成又重新受到人们的关注。常规的DNA 聚合酶具有模板依赖性,无法用于DNA 的从头合成。寻找非模板依赖性的DNA合成酶成为酶法DNA合成技术开发的首要任务。此外,使酶在受控条件下逐个添加指定的核苷酸是酶促寡核苷酸合成技术的另一挑战[24]。
Mackey 和 Gilham[25]使 用 核 苷 酸磷 酸 化 酶(PNPase)将引入2'末端封闭的5'-二磷酸-2'-O-(α-甲氧基乙基)核苷酸偶联到寡腺苷酸引物的3'末端,定序合成了寡聚核糖核苷酸链。Gillam 和Smith[26]采用相同的方法合成了寡聚脱氧核糖核苷酸链。England 和 Uhlenbeck[27]则是首先将连接法用于 DNA 合成,使用 T4 RNA 连接酶将 5',3'-核糖核苷二磷酸底物偶联到引发链的3'末端合成寡核糖核苷酸链。1999 年,T4 RNA 连接酶首次被用于固相酶法 DNA 合成[28]。尽管 PNPase 和 T4 RNA 连接酶能够合成RNA 和DNA,但两者偶联效率低,单轮循环耗时长,用于DNA 合成技术的局限性大于可用性[29]。
Bollum 首先发现末端脱氧核苷酸转移酶(TdT)[30],并于 1962 年提出 TdT 可用于单链寡核苷酸合成[31]。1984 年,Schott 和 Schrade 用 TdT 将dNTPs 添加到不同长度的引发链[32]。研究发现TdT 对四种核苷酸的偏好性差异小、偶联效率高,持续合成和延伸单链DNA 可产生长达8000 nt的均聚物[29-30,33-34]。TdT用于可控酶促DNA合成还需有效的可逆终止方法。使用带有阻断基团的RT-dNTP(RT 为可逆终止子,可在 dNTP 的 3'-OH 或其他位置)为底物[35],通过偶联-去阻断两步循环迭代,有望将TdT 用于长链寡核苷酸的定序合成[24]。具有开发潜力的阻断基团包括氨基、烯丙基、磷酸基团、2-硝基苄基、3'-O-(2-氰乙基)等[29]。2018 年,Keasling 团队[8]另辟蹊径在单分子TdT上用可裂解接头连接单个核苷酸,利用TdT将核苷酸添加到引物链后仍保持与DNA 链的连接,有效地阻止DNA 链的进一步延伸(图2)。裂解接头释放TdT 后,DNA 即可重新进行新一轮的核苷酸添加循环。该方法平均偶联效率可达97.7%,单个循环仅需2~3 min。最近,DNA script公司通过TdT 改造结合阻断基团宣称通过酶法以高达99.5%的偶联效率合成了长达280nt 的寡核苷酸。而Camena Bioscience 所宣传的酶法DNA 合成技术gSynth 的偶联效率更高达99.9%。同时,gSynth在合成300 nt寡核苷酸片段时,产物中全长序列比例达到了85.3%,远高于亚磷酰胺合成法的22.7%。虽然酶法DNA 合成技术至今还未见商业化,但从其所具有的潜力可以预见酶法DNA 合成技术将引领新一轮的DNA合成技术革命。
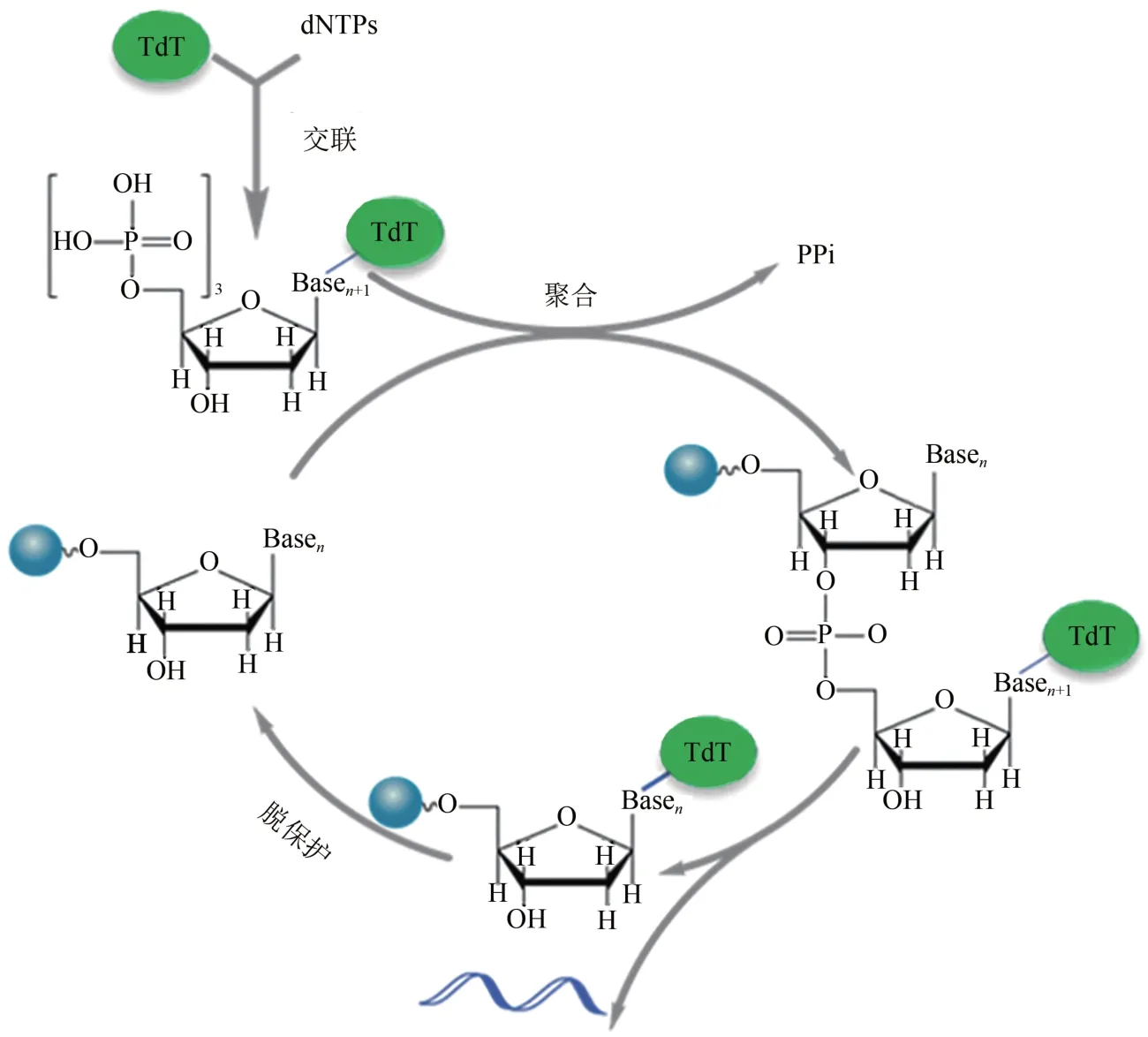
图2 TdT-dNTP 交联体介导的可逆终止用于寡核苷酸合成循环Fig.2 The oligo synthesis cycle by TdT-dNTP conjugates mediated reversible termination
酶作为酶促DNA 合成的关键分子机器,控制聚合物的形成。因此,酶促合成技术的特征与酶功能紧密相关。酶的聚合方式和反应条件温和的特点,使基于酶促的偶联-去阻断两步法循环更为简单,并能有效减少合成过程中发生的DNA 损伤和副反应,有持续合成更长的寡核苷酸片段的潜力。现在DNA 酶促合成技术处于快速发展期,合成过程还存在诸多亟待解决的问题。包括天然酶对修饰RT-dNTP 的掺入效率低、阻断基团的去除、合成无法从头起始或需要一定长度的引发链(如TdT,>3 nt)等。这些问题的核心在于对酶分子机器功能的设计改造和控制策略的开发。通过挖掘新酶、改造酶、研制人工酶等方法提升DNA 合成酶的功能,以高效掺入RT-dNTP;开发新的可控策略适应酶促合成自动化,如通过精细控制辅因子来控制酶活,以提高可控合成效率和长度,也将有助于推动酶促DNA合成的应用。
1.4 DNA合成技术开发评估
DNA 的化学和酶促合成技术的就是根据人为指定的核酸序列,通过相应的合成规则(形成3',5'-磷酸二酯键)从头将核苷酸聚合成寡核苷酸片段。实际得到商业化应用的DNA 合成技术仍限于上述提到的几种。影响这些技术市场化的因素有多种,其中两个主要的技术评估指标是:可控和效率。无模板DNA 合成技术的关键在于可控地逐个添加核苷酸,从而延伸多聚核苷酸链。理论上,当单个核苷酸实现了可控添加时,则全序列即可实现可控合成。在实现可控合成循环后,合成效率和偶联效率将是DNA 合成技术是否具有产业化价值的另一评估指标。合成效率反映在添加单个核苷酸添加循环所耗费的时间(t),柱式法合成一条长度为xnt 的寡核苷酸合成所需总时间T=t×(x−1)。偶联效率则是100 nt 的寡核苷酸分子中的1 个在偶联步骤期间不能反应的概率,可被表述为99%的偶联效率(CE),则全长产物(FLP)=(CE)n,其中n是循环迭代次数(n=x−1)。例如,具有99%CE 的合成200 nt链,单循环为8 min,理论上在1592 min 后产物中只有13%的FLP,87%的为短链或其他错误链。这两个效率与寡核苷酸合成成本紧密相关,也影响更长片段合成的组装和检测筛选过程的耗费。
2 长片段DNA组装技术
目前DNA 片段从头合成的长度有限,更长的基因或基因组则需要通过寡核苷酸片段的酶促组装或体内组装获得。通常使用的寡核苷酸组装方法有两种:连接酶组装法(ligase chain reaction,LCR) 和聚合酶组装法(polymerase cycling assembly, PCA)[6]。连接酶组装法通过DNA 连接酶将首尾相连、重叠杂交的5'磷酸化寡核苷酸片段连接成双链DNA。聚合酶组装法则利用DNA 聚合酶延伸杂交的重叠寡核苷酸片段获得不同长度的混合物,最后用引物扩增出成功组装的全长片段(图3)。PCA 具有良好的兼容性,也被应用于芯片合成的寡核苷酸组装[36-37]。
为了提高基因合成通量并降低成本,整合了合成和组装的微型化和自动化基因合成技术也取得了新进展。2011 年,Tian 等[38]开发了一种采用多功能芯片和组合酶技术的基因合成方法,将整个基因合成过程从寡核苷酸库合成、库扩增、纠错到基因组装等所有步骤整合到同一块芯片上,中途无需更换反应体系,极大地简化了基因合成流程。Twist Bioscience 公司以Agilent 的寡核苷酸原位合成技术为基础,开发了一套对接式硅片反应器用于自动化基因合成。整合了合成和组装的酶促基因合成技术也取得了新进展。据报道,gSynth 酶法DNA 合成技术通过合成与组装的循环实现了2.7 kb的pUC19质粒的从头合成。
对于寡核苷酸组装后双链DNA 的进一步拼接,早期的方法依靠限制性内切酶产生的黏性末端来串联DNA 片段。由此发展出来的有BioBrick与BglBrick 系统以及采用ⅡS 型限制性内切酶切割产生黏性末端实现组装的Golden Gate 技术[39](图3)。但序列依赖性和DNA 残痕的引入以及烦琐的操作过程限制了这类方法的应用。利用核酸外切酶、DNA 聚合酶与连接酶的单独或协同作用开发的组装方法则摆脱了对限制性内切酶的依赖。这类方法通过产生同源单链互补末端进行组装,包括 SLⅠC[40]、SLiCE[41]、LCR[42]、CPEC[43]和Gibson 组装[44](图3)等多种高效简单的组装方法。其中Gibson 组装通过体外一步拼接可以无缝组装长达几十万碱基对的基因组水平的片段。
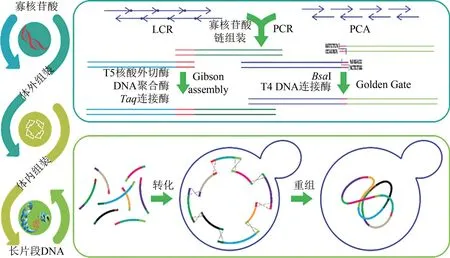
图3 常见体外和体内DNA组装技术及流程Fig.3 Summary of general schemes of in vitro and in vivo DNA assembly
随着片段长度的增加,DNA 在体外很容易受常规操作影响而变得不稳定,超过20 kb 片段的拼接更多借助生物体内的重组系统进行(图3)。大肠杆菌、枯草芽孢杆菌和酿酒酵母是体内DNA 长片段组装的主要宿主细胞[45],经重组系统与细菌人工染色体(BAC)[46]、枯草芽孢杆菌基因组(BGM)[47]或酵母人工染色体(YAC)[48]重组后,这些宿主细胞可稳定携带大片段DNA,其中BGM具有超过3 Mb 的克隆能力[49]。相较于大肠杆菌和枯草芽孢杆菌,酿酒酵母拥有更高的同源重组率,对长片段的兼容性好,是同时装配多个DNA 片段的首选底盘,基于该系统开发的应用方法也更多[50-52]。Gibson 等[53]在酿酒酵母中一步装配 25 个DNA 片段,形成一个长592 kb 的环状支原体基因组。中国科学院的研究人员甚至将接近12 Mb的酿酒酵母完整基因组拼接成单一的染色体[54]。
现有DNA合成技术的局限,使DNA组装成为基因合成不可或缺的过程。组装过程受连接效率和拼接次数影响显著。使用短初始片段组装染色体或基因组长度DNA 所需的分层组装次数较多,过程中所需的克隆挑选和测序等质控成本也会相应增多。此外,组装技术仍面临着一系列问题,需要进一步克服聚合序列、长重复序列和非规范的DNA 结构对组装的抑制作用,开发能稳定得到全长无差错遗传系统的载体等。组装工艺优化或新组装技术的开发有待于对上述组装影响因素的微观认识和有效控制。通过计算机算法对影响组装的问题序列进行拆分和序列转换,发展智能组装技术,将有助于解决复杂长片段DNA 的组装难题。具有低成本、自动化和一体化特性的微流控组装体系将成为寡核苷酸体外合成和组装整合平台开发的方向;而拥有高效重组系统新宿主的筛选与应用将为DNA体内组装提供更多的选项。
3 DNA纠错技术
寡核苷酸合成与酶促组装过程都不可避免地产生多种类型的错误。常见的错误包括核苷酸的插入、缺失和取代。纠错技术的使用可有效地去除不同类型的错误从而提高合成产物的正确率[55]。
3.1 错误寡核苷酸链的去除
经化学合成的寡核苷酸链含有大量错误,对这样的寡核苷酸池的纠错过程主要根据合成错误造成的分子量或基团的差异进行分离纯化。如对柱式合成可通过高效液相色谱法(HPLC)[56]、聚丙烯酰胺凝胶电泳(PAGE)[57]或疏水性纯化柱过滤[10]去除合成不完全的片段。这些方法通量低,对错误区分精确度有限,分离过程损失较大。芯片合成寡核苷酸的纠错可通过直接与序列正确的寡核苷酸捕获探针进行杂交选择[37],或是Tm均一化(热力学参数)严格杂交筛选手段[58],过滤寡核苷酸池中的错误片段。Evonetix 的DNA 合成平台则通过对温度的控制将合成、组装、纠错进行整合,其纠错过程由精确的温度控制去除非完全匹配的DNA 链。还可以使用NGS 技术纠错,结合DNA 芯片合成与高通量测序平台,将合成、组装、测序纠错一体化[59]。
3.2 含错双链DNA片段的纠错
经互补配对后的双链DNA片段的核苷酸插入、缺失、取代错误主要表现为错配和凸起等,这些错误的去除更多是借助基于生物体内的DNA 修复体系开发出的DNA 酶促纠错技术[60-61]。通过对互补序列退火,暴露出错配信号,再利用具有错配结合或错配切割活性的酶对DNA 双链纠错(图4),从而富集正确序列[10,55]。

图4 基于错配切割和错配结合的纠错策略Fig.4 Strategies for error-removal based on mismatch cleavage and mismatch binding
参与DNA修复的错配结合酶MutS及其同源蛋白可以识别并结合各种含错配碱基与单链环的DNA[62-63],然后可通过凝胶电泳、毛细管电泳、亲和磁珠或吸附树脂等方法使MutS 结合含错配的异源双链与未被结合的同质双链分离。这样两轮重复后,可将错误率降低到1/10 kb,与传统的基因合成技术相比错误降低超过15 倍[64-65]。针对错配结合酶MutS 的工程改造,在提其高稳定性的同时也有助于市场化应用[66]。对于错误率高的寡核苷酸池,Binkowski 等[67]利用 MutS 进一步开发了同序改组法,通过引入限制性内切酶对DNA 双链进行片段化,使得MutS 不需要去除整条错误的双链DNA,从而保留大量含有正确序列的短片段,最后通过OE-PCR 组装回收全长序列。测试发现3.5 kb 的片段经过两轮同序改组后错误率降低至1/3.5 kb,正确率提高了3.5~4.3倍。MutS纠错方法本质是对含错DNA 双链的物理分离。为保证MutS处理后样品中有足够的正确片段,寡核苷酸池中需要有相当一部分序列正确的片段。
运用错配切割酶能达到在原处理池中纠正错误的目的。错配切割酶是识别DNA 双链错配位点并在错配位点附近切割的一组错配特异性核酸内切酶。主要包括识别单碱基错配的核酸内切酶和单链特异性核酸酶[68-70],这些酶与聚合酶共同作用,利用具有核酸外切酶活性的聚合酶水解切割错误区域,然后通过OE-PCR 组装回收全长序列。这种方法可以消除单碱基水平的错误,同时保留大部分序列正确的区域,并且可以进行多次“纠错-组装”循环,直到获得所需纯度的产品。T4 核酸内切酶Ⅶ,T7 核酸内切酶Ⅰ和E.coli核酸内切酶Ⅴ等可识别并切割双链DNA 中单碱基错配、单核苷酸凸起等类型的错误,能有效减少合成基因产物中错配、缺失和插入等错误[69,71-73]。单链特异性核酸酶中CEL 可在中性pH 值下特异性切割不同类型的碱基错配和DNA 畸变[74-76]。在芯片的基因合成纠错中结合使用CEL 核酸酶可以将合成基因产物的错误率从1/526 bp减少到1/3883 bp。两次酶解错误切割反应可进一步将错误率降低至1/8700 bp,错误减少了16倍以上[77]。
现有DNA 纠错方法大部分还停留在对含错片段的去除,开发基于错配切除并对错误进行修复的纠错方法有望颠覆传统的纠错技术。错配修复需要正确的模板链,体内的错配修复系统通过甲基化修饰区分模板链与新合成子链,并根据模板链来修复新合成链上的错误。借鉴类似的原理在体外区分正确片段与待修复片段,在此基础上识别错误并对错误区域进行单链切割,进而以正确片段为模板修复错误。以此开发的纠错技术将摆脱传统纠错技术的局限,实现真正的体外DNA纠错。
4 结 语
寡核苷酸合成、组装与纠错三个过程的核心目标都是核酸。从单核苷酸到寡核苷酸,从短片段到长片段,从高错误率到低错误率的目标,最终获得准确序列的长片段DNA。通过不同的酶分子机器来实现各过程的目标,进而相互补充以达到更高目标。在以酶为核心的DNA 酶促合成中,酶作为实际过程的首要执行者,其功能直接影响最终产物的质量。然而用于不同阶段的酶分子机器在反应机制和控制技术上差异巨大。自然界筛选获得的酶分子机器又无法直接满足DNA 合成应用的需求,这就要求我们对相应的酶进行改造。按设计方法去找寻甚至设计新酶,或依赖酶去设计新方法这两种方案的精细化控制,也将伴随着其他技术的进步,成为高效DNA合成的重要方法。
近年来,DNA 合成、组装与错误纠正技术的不断发展,使染色体或基因组的合成、人工设计基因组的创造、可控细胞工厂与人工生物的构建都成为可能[78-82]。这些研究也推动了合成生物学的快速发展,其中,设计构建新功能基因、遗传网络甚至基因组,以实现从头合成、按需合成和生物大分子的定向改造的时代已经到来。各界对高效保真的DNA 合成技术,高通量的组装与错误校正体系开发等基因合成相关技术的需求更为强烈。DNA 合成技术在生物医疗、生物制造、DNA 存储等诸多领域具有广阔应用前景与巨大的市场潜力[83-84],预计 2030 年全球 DNA 合成市场将增长到1.6万亿美元。然而2018年11月,合成生物学被美国列为拟限制出口的前沿生物技术领域之一,高性能DNA 合成仪已禁止向中国销售,这将严重影响我国合成生物学的健康发展。因此,推动DNA合成、组装、纠错等技术的发展,开发长片段高效无差错的微量DNA 合成技术,利用计算机设计序列优化组装过程,结合酶促或NGS 测序技术开发DNA 合成纠错平台,以绿色、高通量、自动化和一体化的方式低成本高质量地合成DNA,实现大规模基因和基因组合成,具有重要科学意义和重大应用价值。
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
肝博士(2022年3期)2022-06-30
作物学报(2022年7期)2022-05-12
化学研究(2021年4期)2021-08-19
井冈山大学学报(自然科学版)(2021年1期)2021-03-05
海外星云 (2021年21期)2021-01-19
中国药科大学学报(2020年1期)2020-05-12
中国饲料(2019年19期)2019-03-25
智富时代(2018年9期)2018-10-19
