
多变鱼腥藻ATCC 29413基因的一步插入失活
2021-02-04 06:39雷棋琴戢水玲黄任衡徐旭东孔任秋
水生生物学报 2021年1期
雷棋琴 戢水玲 黄任衡 高 宏 徐旭东 孔任秋
(1.中国科学院水生生物研究所中国科学院藻类生物学重点实验室,武汉 430072;2.中国科学院大学,北京 100049)
多变鱼腥藻(Anabaena variabilis)ATCC 29413是一种淡水丝状蓝藻,在缺氮条件下藻丝中一些间隔分布的营养细胞分化为异形胞进行固氮;在干旱等极端环境下还可以分化形成厚壁孢子。该藻株不仅可以利用光合作用进行自养生长,也能利用果糖作为碳源和能源在完全黑暗条件下异养生长;其最适生长温度为30℃,但在40℃高温条件下生长状态良好,属于中等嗜热型藻[1]。利用丝状蓝藻的接合转移系统,多变鱼腥藻ATCC 29413高温衍生株可以进行遗传操作,利用同源重组得到特定基因的突变株[2—6]。
具有异养生长能力的蓝藻依靠单糖或双糖作为碳源[7—10],在光合作用、光合膜的发生等有关基因被失活时仍然能够生长,为研究这些基因的功能提供了便利;作为光合细胞工厂,用于生产各种高价值化学品和生物燃料,也具有一定优势[11]。然而,多变鱼腥藻ATCC 29413的接合转移效率很低,影响了研究工作的开展。我们对该藻株已发表的基因组序列进行分析发现,其含有两套Ⅱ型限制修饰系统,包括AvaⅠ/M.AvaⅠ和AvrⅡ/M.AvrⅡ。这两种限制酶的存在可切断进入细胞的外源DNA,妨碍接合子的形成。过去所用的丝状蓝藻接合转移系统中的辅助质粒pRL623上缺少甲基化酶M.AvrⅡ基因,可能与难以构建多变鱼腥藻突变株有关[12,13]。为了克服藻株的限制酶对外源DNA的破坏,本研究克隆了该藻株自身的两个甲基化酶基因,构建了新的辅助质粒。利用该质粒对多变鱼腥藻ATCC 29413两个基因进行了插入失活,均实现了一步获得同源双交换子,比通常在丝状蓝藻敲除基因节省一半时间。
1 材料与方法
1.1 藻种、菌种及培养方法
多变鱼腥藻ATCC 29413高温衍生株由美国南达科塔州州立大学周阮宝教授惠赠。藻株接种在稀释8倍的AA液体培养基(AA/8)[14]或BG-11固体培养基[15]生长,接合转移时固体培养基中添加5 mmol/L的果糖,突变藻株的培养基中补加10 μg/mL的红霉素(Em)。在(30±1)℃、30 μE/(m2·s)持续光照条件下静置培养,通过测定A730来监测其生长,待藻细胞到达对数期时用于实验。大肠杆菌(Escherichia coli)在37℃的LB培养基中培养,含有质粒的大肠杆菌根据所带抗性基因补加50 μg/mL壮观霉素(Sp)、氨苄青霉素(Ap)、卡那霉素(Km)、10 μg/mL的氯霉素(Cm)或10 μg/mL四环素(Tc)。
1.2 蓝藻基因组提取和PCR反应
蓝藻基因组提取和PCR反应参照文献[16],引物合成和序列分析在武汉天一辉远生物科技有限公司进行。
1.3 质粒的构建
DNA重组按Molecular Cloning[18]描述的方法进行。限制性内切酶、连接酶、质粒pMD18-T购自大连宝生物工程有限公司。质粒pHB518(GenBank登录号EF688562.1)和pHB576(GenBank 登录号EF688561.1)是本实验室前期构建的载体。pDS4101[2]、pRL443[2]、pRL623[13]、pRL277(GenBank 登录号L05082.1)和pACYC184(GenBank 登录号X06403.1)由美国密歇根州立大学Peter Wolk教授惠赠。
辅助质粒的构建根据多变鱼腥藻ATCC 29413限制酶的甲基化酶基因M.AvaI和M.AvrII(ava_3181和ava_4359)的DNA序列设计两对引物(表1),两个基因的上游引物序列在ATG前7个碱基设计典型核糖体结合位点AGGAGG,M.AvrII基因的上游引物设计上终止密码子用于终止插入载体pACYC184上四环素抗性基因的翻译。以野生型多变鱼腥藻ATCC 29413基因组为模板,以引物M.AvaI p-1/p-2和M.AvrII p-1/p-2分别进行PCR扩增,得到大小为1.45 kb的M.AvaI基因和1.28 kb的M.AvrII基因,经过纯化连接到pMD18-T载体上,转化E.coliDH10B,得到质粒pHB6065和pHB6067,感受态菌制作方法参照文献[19]。将质粒pHB6065用BamHⅠ酶切补平后再用SphⅠ酶切,琼脂糖电泳回收1.45 kb的M.AvaI片段,同时将质粒pHB6067用SalⅠ酶切补平后再用SphⅠ酶切,回收线性化4 kb大小带M.AvrII的载体片段。将目的片段与载体连接后转化感受态E. coliDH10B,得质粒pHB6087;将pHB6087质粒上2.7 kb的BamHⅠ(补平)和SalⅠ双酶切片段(含有M.AvaI和M.AvrII)克隆到质粒pACYC184的EcoRV和SalⅠ双酶切位点,得到辅助质粒pHB6088。所有构建的质粒经PCR检测和测序验证无误。
用于基因插入失活的运载质粒的构建根据已公布的基因ava_1237和ava_4412序列发现两基因大小分别为1.42和0.48 kb。为了便于同源交换,在每个基因两侧分别设计了两对引物(表1),以多变鱼腥藻基因组DNA为模板,利用引物ava_1237 p-1/p-2扩增出基因ava_1237上游0.71 kb的片段,以引物ava_1237 p-3/p-4扩增出基因ava_1237下游0.70 kb的片段,将两片段分别克隆到质粒pHB518和pHB576的Eam1105Ⅰ位点,同时删除了以上两个质粒上的氯霉素抗性基因(Cmr)得到质粒pHB5225和pHB5235,用NotⅠ酶切pHB5235,插入从pHB5225中用NotⅠ酶切回收的约2.7 kb片段(含有ava_1237上游片段+Emr);通过PCR鉴定,选择上下游片段方向一致的克隆,得到质粒pHB5245;用PvuⅡ酶切pHB5245,回收约3.7 kb片段(含有ava_1237上游片段+Emr+ava_1237下游片段),将其插入到pRL277质粒PstⅠ位点(PstⅠ酶切并补平)得到运载质粒pHB5255。
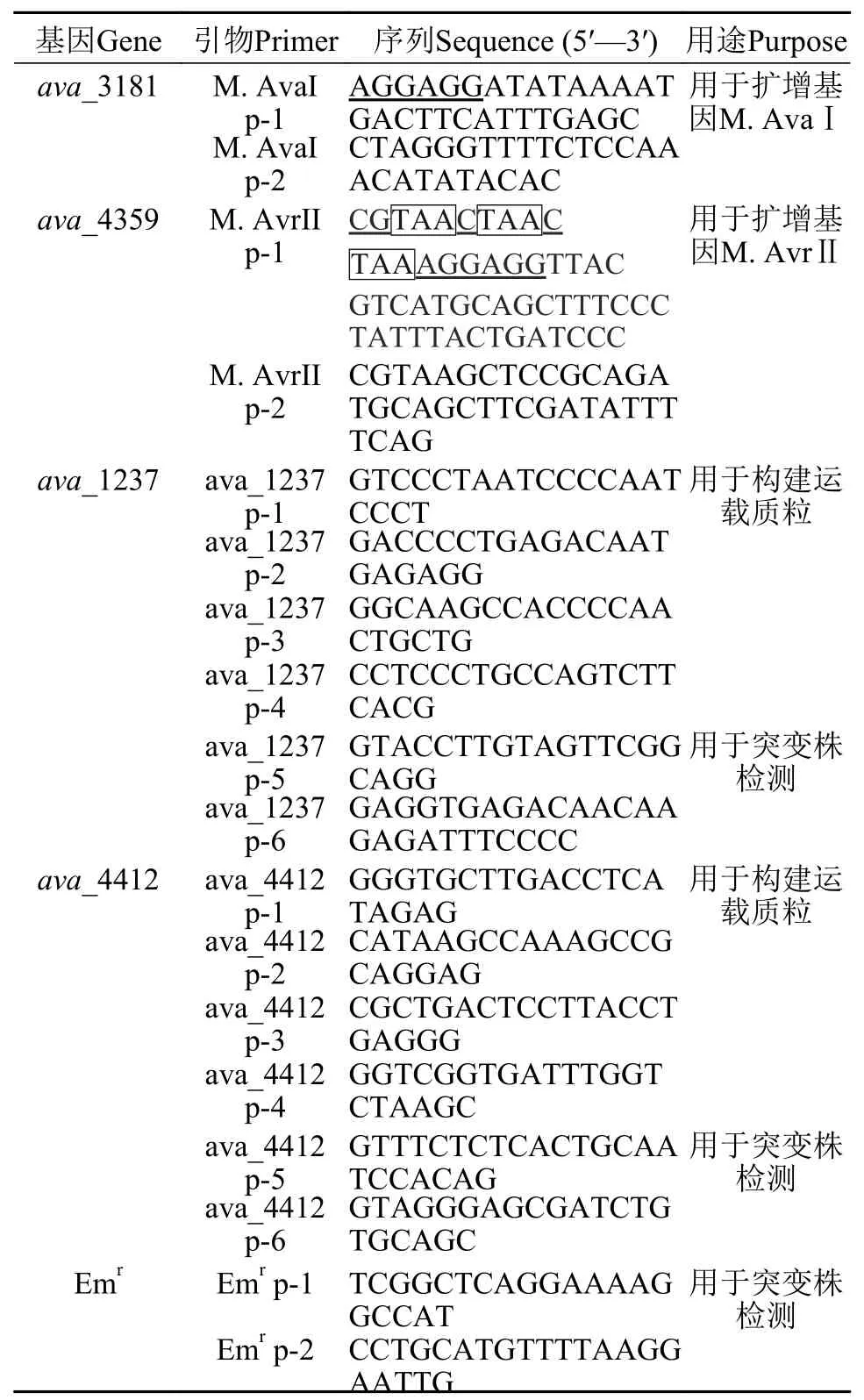
表1 本文所使用的引物Tab.1 Primers used in this study
同样的以多变鱼腥藻基因组DNA为模板,利用引物ava_4412 p-1/p-2扩增出基因ava_4412上游0.69 kb的片段,以引物ava_4412 p-3/p-4扩增出基因ava_4412下游0.86 kb的片段,将两片段分别克隆到质粒pHB518和pHB576的Eam1105Ⅰ位点,同时删除了以上两个质粒上的氯霉素抗性基因得到质粒pHB5231和pHB5241;用Sse8387Ⅰ酶切pHB5241,插入从pHB5231中用Sse8387Ⅰ酶切并回收的约2.6 kb片段(含有ava_4412上游片段+Emr);通过PCR鉴定,选择上下游片段方向一致的克隆,得到质粒pHB5251;用PvuⅡ酶切pHB5251,回收约3.7 kb片段(含有ava_4412上游片段+Emr+ava_4412下游片段),将其插入到pRL277质粒PstⅠ位点(PstⅠ酶切并补平),得到运载质粒pHB5261。
1.4 接合转移菌株的构建
在接合转移前需要把几种质粒转入同一个菌株,其中包括辅助质粒、运载质粒和接合质粒。首先通过转化将辅助质粒和运载质粒转入同一个大肠杆菌细胞E. coliDH10B,然后将含有接合质粒的大肠杆菌细胞与含有辅助质粒和运载质粒的大肠杆菌细胞在含有相应抗生素的LB培养基上交叉划线,长出的细胞即是含有辅助质粒、运载质粒和接合质粒的菌株。
辅助质粒pHB6088转化到E. coliDH10B(pDS4101)细胞中,得到菌株E. coliDH10B(pDS4101+pHB6088)。再将两个运载质粒pHB5255和pHB5261分别转化到感受态细胞E. coliDH10B(pDS4101+ pHB6088)中,得到含3种质粒的菌株E. coliDH10B(pDS4101+pHB6088+运载质粒)。用接种环将上述含3个质粒的菌株与E. coliHB101(pRL443)在含有Ap、Cm、Sp、和Tc四种抗性的固体LB平板上交叉划线,质粒pRL443自主转移到含3质粒的菌株中,得到含4种质粒的菌株E. coliDH10B(pDS4101+pHB6088+运载质粒+pRL443)。另外将辅助质粒pRL623转化到含有运载质粒的细胞中,然后再与含有pRL443的菌株细胞交叉划线,在三抗培养基(Cm、Sp和Tc)上筛选含有pRL623、运载质粒和pRL443的大肠杆菌细胞,得到E. coliDH10B(pRL623 +运载质粒+pRL443)。
1.5 接合转移和突变株的筛选
接合转移方法参照文献[2]采用两个途径进行,分别用菌株E.coliDH10B(pDS4101+pHB6088+运载质粒+pRL443)和菌株E.coliDH10B(pRL623+运载质粒+pRL443)与多变鱼腥藻ATCC 29413进行接合转移,按比例取50 mL对数生长期藻细胞(A730为0.4)和25 mL大肠杆菌接合转移细胞(A600为0.6),先将藻和菌分别用无抗的培养基洗3遍,再将藻和菌分别浓缩为1 mL,藻菌混合液涂于带硝酸纤维素滤膜的无抗BG11平板(添加5% LB),每个BG11平板涂100—200 μL的藻菌混合液,恢复一天后再从滤膜上洗下藻菌混合物,1800 r/min离心收集藻细胞后,将其涂于添加红霉素和5 mmol/L果糖的固体BG11平板上等待接合子长出。长出的接合子在含红霉素的BG11平板上划线培养,待长出来之后转入红霉素抗性的30 mL AA/8液体培养基,根据不同的基因设计检测引物,传代分离纯化后提取野生型和接合子基因组DNA做PCR检测。
2 结果
2.1 用于甲基化保护的辅助质粒

图1 辅助质粒构建流程图Fig.1 Schematic diagram of construction of the helper plasmid
辅助质粒的构建流程如下(图1):将多变鱼腥藻ATCC 29413中限制酶(AvaⅠ和AvrⅡ)的甲基化酶基因ava_3181(M. AvaⅠ)和ava_4359(M. AvrⅡ)分别克隆到pMD18-T载体上,得到质粒pHB6065和pHB6067。然后将质粒pHB6065上的M. AvaⅠ基因以相同方向克隆到pHB6067质粒M. AvrⅡ基因下游得到pHB6087。最后将串联的M. AvaⅠ和M.AvrⅡ插入到质粒pACYC184四环素抗性基因中间,得到辅助质粒pHB6088。pHB6088利用四环素抗性基因的启动子来驱动M. AvrⅡ和M. AvaⅠ的转录(图1),基因M. AvrⅡ上游设计带有终止密码子(Stop codon)和核糖体结合位点(RBS),Stop codon用于终止Tcr上游序列的翻译,RBS为大肠杆菌典型核糖体结合位点(AGGAGG)。
2.2 用于基因敲除的运载质粒
基因ava_1237的侧翼同源序列0.71和0.7 kb分别克隆到质粒pHB518和pHB576得到质粒pHB 5225和pHB5235,再将pHB5225的上游侧翼同源序列连同Emr切下连接到pHB5235上得到质粒pHB5245,质粒pHB5245上的两个侧翼同源序列和Emr切下连到载体pRL277上得到运载质粒pHB5255。
基因ava_4412的侧翼同源序列0.69和0.86 kb分别克隆到质粒pHB518和pHB576得到质粒pHB 5231和pHB5241,再将质粒pHB5231的上游侧翼同源序列和Emr切下连接到pHB5241上得到质粒pHB5251,质粒pHB5251上的两个侧翼同源序列和Emr切下连到载体pRL277上得到运载质粒pHB5261。
运载质粒pHB5255含有基因ava_1237上下游两侧分别约有0.7 kb的序列,其基因中间142 bp的序列被Em抗性基因置换,用于ava_1237基因失活研究;另一个运载质粒pHB5261含有ava_4412上游0.69 kb和下游0.86 kb序列,其基因中间126 bp的序列被Em抗性基因置换,用于ava_4412基因失活研究。两个运载质粒上面还含有bom位点和用于双交换筛选的蔗糖致死基因(sacB)及壮观霉素抗性基因(Spr)。
2.3 基因的一步插入失活
本研究构建了两种接合转移菌株E.coliDH10B(pDS4101+pHB6088+运载质粒+pRL443)和E.coliDH10B(pRL623+运载质粒+pRL443)。pDS4101提供接合转移所需的Mob蛋白;pHB6088含有多变鱼腥藻两个甲基化酶基因(M. AvaⅠ和M. AvrⅡ),对运载质粒特定位点进行甲基化保护;pRL623上有来源于其他微生物的三种甲基化酶基因(M.AvaⅠ、M. Eco47Ⅱ和M. EcoT22Ⅰ)和Mob蛋白基因;接合质粒pRL443,具有自主转移特性并提供接合所需的性纤毛,质粒上的tra基因丛编码的蛋白能驱动运载质粒从E. coli到藻细胞的接合转移;运载质粒含bom位点,克隆有本研究将要敲除的基因的上下游同源序列和插入其间的红霉素抗性基因。
将以上菌株分别与多变鱼腥藻ATCC 29413进行接合转移后,在添加果糖和红霉素的BG11平板上筛选。7—14d后,利用含有辅助质粒pHB6088的菌株进行的接合转移平板开始长出接合子,每组都长出20个左右的接合子,而利用含有辅助质粒pRL623的菌株进行接合转移的平板没有接合子长出。将接合子划线到添加果糖和红霉素的BG11固体平板培养,长出的藻落再接种到添加果糖和红霉素的AA/8液体培养基中传两代。提取基因组DNA,在基因组与运载质粒同源序列的上下游两侧和红霉素抗性基因内部设计引物(表1),进行PCR检测与比较。这两个突变株分别随机检测了2个(ava_1237)和4个接合子(ava_4412),都显示发生了同源双交换,而不是单交换(图2),也就是省略了在丝状蓝藻构建基因敲除突变株要先获得单交换株的传统实验步骤。图2中泳道3和4都显示获得了基因ava_1237的双交换突变,但泳道3检测的接合子突变尚未完全分离(蓝藻基因组有多拷贝,该接合子仍有野生型拷贝);泳道7和9是基因ava_4412完全分离的双交换突变株,泳道8和10是未完全分离的突变株;泳道1、2和5、6是两个基因的野生型对照。图3为同源双交换示意图。

图2 突变株PCR检测结果Fig.2 PCR examination of mutants
由于以PCR随机检查的6个接合子全部为双交换株,本实验室敲除其他基因获得的几十个接合子也均为双交换株,我们推测以该遗传操作系统获得的全部或绝大多数接合子均直接发生双交换。因此,可按所得到的抗性接合子数除以藻细胞数计算通过接合转移敲除基因的效率。结果显示,用辅助质粒pHB6088进行接合转移,获得基因ava_4412插入失活突变株的效率约为4.95×10-8,基因ava_1237插入失活的效率约为4.09×10-8,而以pRL623作为辅助质粒进行接合转移未得到接合子。

图3 同源双交换示意图Fig.3 Schematic diagram of homologous double crossover
3 讨论
在丝状蓝藻中,通常利用接合转移的方法来开展遗传操作[2],但接合转移仅仅提供一个外源DNA导入藻细胞的途径,DNA进入后能否在细胞内复制或整合还受多种因素的影响,尤其是藻株的限制修饰系统(R-M系统)[12]。为了克服限制酶对外源DNA的降解,采用较多的方法是在导入蓝藻之前对DNA进行甲基化修饰从而逃脱蓝藻限制酶的切割[20]。因此在辅助质粒上可克隆藻株的甲基化酶基因,在大肠杆菌对运载质粒进行甲基化保护,从而提高接合转移的效率。比如用于鱼腥藻PCC 7120接合转移的辅助质粒pRL623上就有三种甲基化酶基因(M. AvaⅠ、M. Eco47Ⅱ和M. EcoT22Ⅰ),针对其三种限制酶(AvaⅠ、AvaⅡ和AvaⅢ)进行甲基化保护,极大地提高了接合转移效率[13]。本研究发现,应用丝状蓝藻辅助质粒pRL623在对多变鱼腥藻ATCC 29413接合转移时得不到接合子,通过克隆多变鱼腥藻ATCC 29413基因组中的两个甲基化酶基因(ava_3181和ava_4359),构建辅助质粒pHB6088,结果获得了接合子,相对而言在一定程度上促进了遗传操作的成功。此外,利用这个接合转移系统和辅助质粒pHB6088,我们又进行了19个未知功能基因敲除或失活并获得了成功。因此,辅助质粒pHB6088比pRL623更有利于对多变鱼腥藻ATCC 29413进行遗传操作。
有研究表明在蓝藻细胞中提取的质粒DNA由于被甲基化而不能转化基因型为mcrA+的E. coliDH5a[21],这是由于在一些大肠杆菌株系中含有特异性降解甲基化DNA的限制酶系统[22,23]。本研究构建的辅助质粒在尝试转入E. coliDH5α(mcr+mrr+)菌株时无法得到转化子,而转入E. coliDH10B(mcr-mrr-)没有障碍,这也说明辅助质粒上的甲基化酶基因有表达,发挥了甲基化保护作用。
在蓝藻的接合转移中,一般运载质粒进入藻细胞后需要经历单交换和双交换两个过程的筛选[24],现有文献报道的对多变鱼腥藻ATCC 29413基因敲除的研究也是要先发生单交换[4—6]。但作者在基因敲除的过程中观察到,多变鱼腥藻ATCC 29413在某种条件下,可一步发生同源双交换,直接跳过单交换筛选得到插入失活突变株,这在一定程度上缩短了获得突变株的周期。通常接合转移效率是依据导入能自主复制的质粒获得接合子的数量来计算的,而本研究导入的是不能复制的质粒,进入细胞后一步发生双交换,所以获得接合子的频次不高。以一步发生双交换的概率来看,本文所构建的接合转移系统效率并不低。
我们对多变鱼腥藻ATCC 29413基因组序列搜索比较发现,该藻株也含有识别和切割特定位点甲基化DNA的Mrr限制系统(由ava_B0091基因编码)[23,25]。推测如果运载质粒上有Mrr识别位点而目的基因侧翼片段和抗性基因上没有,很可能进入藻细胞的DNA质粒被部分切割、线性化,导致基因侧翼片段直接与目标基因进行同源双交换。因此,为了进一步提高该藻株的接合转移效率,构建Mrr限制系统缺陷藻株,消除Mrr限制系统对DNA的影响也是有必要的。
猜你喜欢
河北科技师范学院学报(2022年2期)2022-08-26
成都医学院学报(2022年4期)2022-08-19
基层中医药(2022年4期)2022-07-22
汉字汉语研究(2021年2期)2021-08-30
空间科学学报(2021年1期)2021-05-22
江西农业学报(2021年4期)2021-04-20
三农资讯半月报(2020年11期)2020-06-21
汉字汉语研究(2019年2期)2019-08-27
新高考·英语进阶(高二高三)(2018年8期)2018-01-15
中国果菜(2016年9期)2016-03-01
