
基于高通量测序的菟丝子生防菌“鲁保一号”转录组学研究
2021-02-07 01:24李健李美高兴祥房锋
山东农业科学 2021年1期
李健,李美,高兴祥,房锋
(山东省农业科学院植物保护研究所,山东 济南 250100)
菟丝子是菟丝子属(Cuscuta sp.)植物的统称,能够通过吸器汲取寄主的养分,是危害严重的一类恶性寄生性杂草。近些年,菟丝子的侵入不仅对大豆等作物造成了危害,而且对我国甘肃、宁夏、内蒙古和辽宁等地的草原牧区生态造成了严重威胁[1-3]。尖孢炭疽菌“鲁保一号”[3]是山东省农业科学院刘志海等人于1963年研制成功的、一种对菟丝子有特殊防效的微生物菌[4-6]。该菌曾在我国多个省、区得到推广和应用,对控制菟丝子的蔓延和危害起到了一定作用[4,5]。由于该菌株致病力易发生退化,且应用较早,基因资源信息匮乏,严重影响了对其进一步的研究和应用[7]。因此,系统研究“鲁保一号”菌株的转录组信息,挖掘相关功能基因,对解决其培养过程中的致病力退化问题至关重要。
转录组是指细胞在特定状态下表达的全部RNA的总和,反映了相应物种在一定状态下的基因表达状况。由转录组延伸而来的差异转录组能够反映相同基因在不同条件下表达水平的差异,为揭示不同基因的相互调控模式及各自功能提供了可能[8,9]。随着测序仪器的改进和测序原理的明晰,目前新一代转录组测序(高通量测序,highthroughput sequencing)技术已经得到了越来越广泛的应用[10,11]。该技术可以在芯片上并行对数百万计的DNA分子进行大规模测序,从而获得海量的测序结果,结合相应分析手段,使得对缺乏基因组信息的物种进行细致全貌的分析成为可能,是对传统测序技术的一次变革[12-14]。
“鲁保一号”菌株具有较高的应用价值,但是由于其开发时间较早,且受制于其菌种致病力退化,对该菌株的研究基本停止,而对其功能基因信息相关研究则未见报道。对于尚未系统开展基因组学研究的物种来说,获得大通量的基因资源信息是解决问题的首要步骤,新一代高通量测序技术的发展为从分子生物学水平研究“鲁保一号”菌株提供了便利。本试验首次将高通量测序技术应用到“鲁保一号”菌株转录组研究中,对测序获得的海量数据进行拼接与组装,结合生物信息学技术对所获得的unigene进行功能注释和功能分类;以测序获得的数据信息为依据,筛选CDC(细胞分裂周期基因,cell division cycle gene)系列相关基因,并系统分析其遗传进化关系,为“鲁保一号”菌株基因功能的研究奠定基础。
1 材料与方法
1.1 供试材料
试验于2015年12月—2017年5月在山东省农业科学院植物保护研究所杂草科学实验室开展。供试菌株为“鲁保一号”,保存于山东省农业科学院植物保护研究所杂草科学研究室。
培养基为马铃薯葡萄糖培养基(PDA)和完全培养基(CM)。
1.2 转录组测序
PDA培养基活化复壮后的“鲁保一号”菌株记为LB-1,连续继代(培养皿内生长7 d为一代)培养10代后的致病力减弱菌株记为LB-1-10。挑取菌落边缘长势一致的菌丝,接种于液体CM培养基内,160 r/min、黑暗摇培72 h,双层纱布过滤收集菌丝,菌丝1∶1混合后经液氮速冻,送至北京百迈克生物科技有限公司进行RNA的提取与转录组学分析。获得高质量的原始测序数据后,通过Trinity组装软件[15]对相应序列进行组装拼接。首先将测序reads打断为较短的片段(kmer),然后将这些小片段通过序列拼接组装成较长的重叠群(contig),并利用这些片段之间的重叠,得到片段集合(component),最后利用de-Bruijn图的方法和测序read信息,在各个片段集合中分别识别转录本(transcript)序列,对转录本进行同源聚类和拼接得到单基因簇(unigene)。
1.3 功能注释与分类
使用BLAST软件[16]将测序获得的unigene序列与NCBI的非冗余核酸数据库(non-redundant protein database,NR)、Swiss-Prot(swissprot protein sequence database)和蛋白质直系同源数据库(cluster of orthologous groups,COG)[17-19]等蛋白质数据库进行比对分析,获得最佳功能注释。之后使用HMMER软件[20]与Pfam[21]数据库比对,获得unigene的注释信息。根据NCBI数据库的功能注释信息,使用GO软件[22]得到unigene的GO条目,然后用WEGO软件[23]进行分类统计。数据分析过程中,选择BLAST参数E-value不大于1e-5和HMMER参数E-value不大于1e-10。
1.4 CDC相关基因的筛选和进化分析
前期研究显示,细胞分裂紊乱是“鲁保一号”菌株连续继代培养后的重要现象[7]。根据对转录组数据的功能注释结果,结合比对分析筛选获得了6个CDC相关基因。利用MEGA 6软件对获得的相关基因进行系统进化树分析。
2 结果与分析
2.1 “鲁保一号”菌株转录组数据的组装
基于边合成边测序(sequencing by synthesis,SBS)技术,使用Illumina Hiseq 2500高通量测序平台对“鲁保一号”菌株完成转录组测序工作。共获得总长度为431 911 195 bp的序列信息,进一步组装获得10 013 398个contig序列,主要以长度为200~300 bp的contig序列为主,有9 962 103条,占总体的99.49%;300~500 bp的contig序列有22 853条,占总体的0.23%;500~1 000 bp的contig序列有14 422条,占总体的0.14%;1 000~2 000 bp的contig序列有7 677条,占总体的0.08%;≥2 000 bp的contig序列有6 343条,占总体的0.06%(表1)。
对所获得的contig数据进行进一步组装,得到总长度为61 674 287 bp的transcripts,共25 588条,其N50为4 038 bp,组装完整性较高。长度200~300、300~500、500~1 000、1 000~2 000 bp和≥2 000 bp的transcripts序列分别占总体的9.27%、10.70%、15.82%、21.90%和42.30%(表1)。
对获得的transcripts序列进行进一步组装,得到17 031条unigenes序列,总长度为31 126 662 bp,平均长度为1 827.65 bp,N50长度为3 093 bp。长度为200~300、300~500、500~1 000、1 000~2 000 bp和≥2 000 bp的unigene序列分别占总体的12.87%、13.47%、18.22%、22.99%和32.45%(表1)。
2.2 unigene的功能注释、分类和代谢途径分析
将拼装得到的unigene序列与多个公共数据库进行比对,其中KOG数据库中共有5 228个unigene获得注释,GO数据库中共5 192个,NR数据库中共9 991个(表2)。共获得10 538个有注释信息的unigene序列,占全部unigene序列的61.9%。

表1 “鲁保一号”菌株转录组数据的组装统计
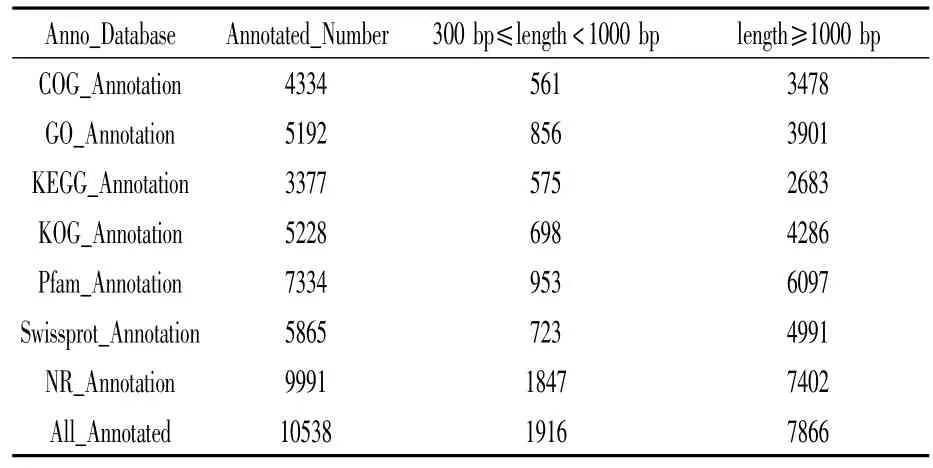
表2 BLAST比对公共数据库结果
2.2.1 unigene的GO注释结果 GO(基因本体,gene ontology)是一个被广泛应用的标准化基因功能分类数据库,数据库分类注释结果总共有三大类,分别是分子功能(molecular function)、细胞组分(cellular component)和生物学过程(biological process)。本试验结果表明,可将转录组获得的所有unigene划分为52个功能组,其中3 754个属于细胞组分,7 935个属于分子功能,7 410个属于生物学过程。其中细胞成分、细胞器成分、催化活性、结合活性、代谢进程、细胞进程和单一生物进程涉及的unigene较多,而病毒体、胞外基质、金属伴侣蛋白活性、通道调节活性和细胞杀伤等涉及的unigene没有或极少(图1)。
2.2.2 unigene的NR注释结果 使用BLAST软件将unigene序列与NR数据库比对,进行序列相似性分析,得到与给定unigene具有最高序列相似性的蛋白描述,并获得unigene蛋白的功能注释信息。由图2可知,86.04%的序列与已知炭疽菌序列有不同程度的同源性,相似序列匹配的近缘物种还有大豆、高粱、西瓜等,其他物种占13.05%。
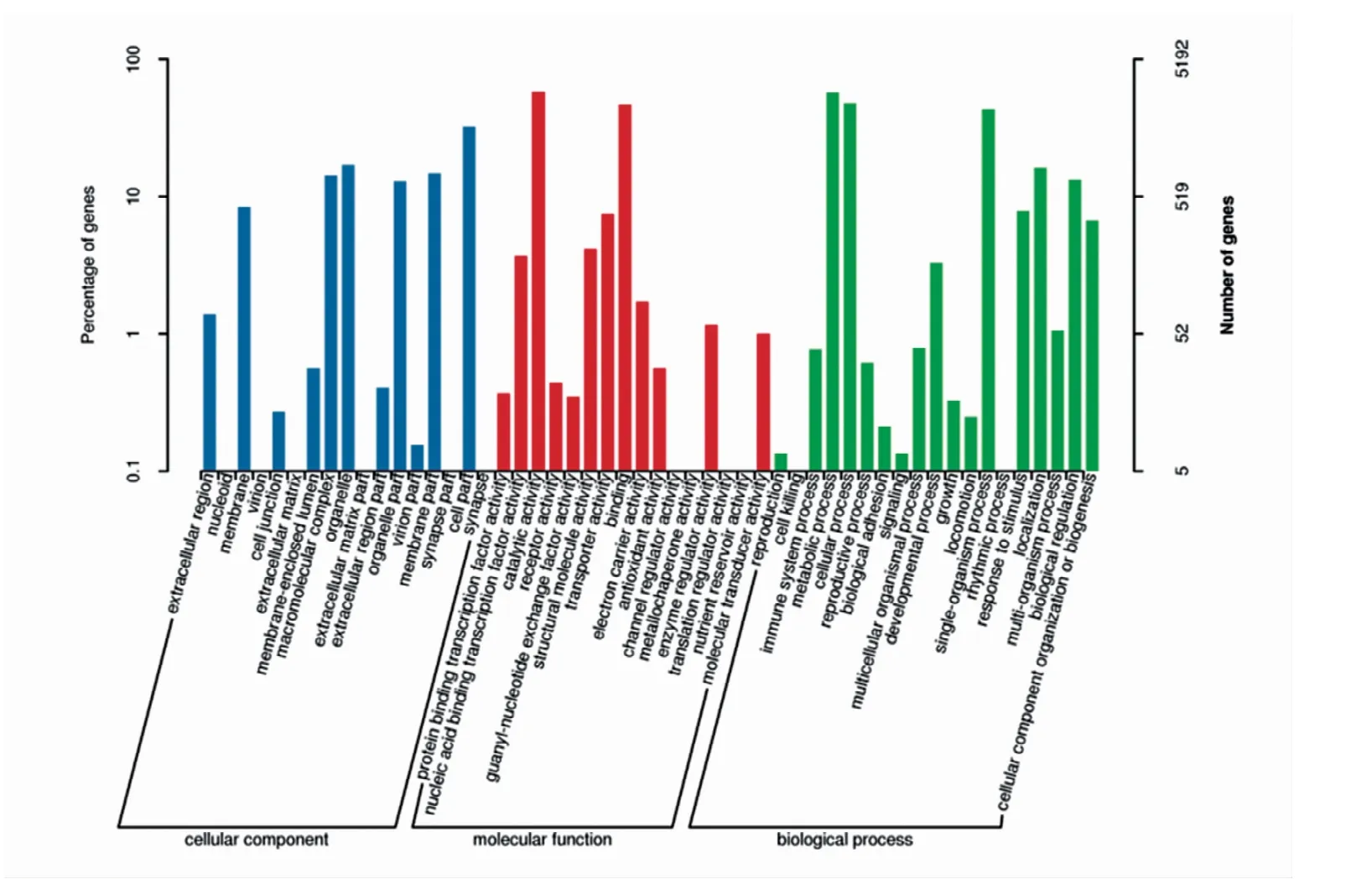
图1 unigene的GO分类结果

图2 “鲁保一号”菌株的同源物种分布
2.2.3 unigene的KOG注释结果 “鲁保一号”菌株的unigene根据其功能大致分为25类(图3),涉及了大多数生命活动,如RNA加工与修饰,染色体结构和动力学,能量产生与运输,细胞周期控制、细胞分裂及染色体分裂,氨基酸运输及代谢等。其中注释最多的是一般功能预测类基因(R),其次是翻译后修饰、蛋白折叠和分子伴侣类基因(O),再者是翻译、核糖体结构和生物发生类基因(J),只有极少数的细胞活性类基因(N)和胞外结构类基因(W)。
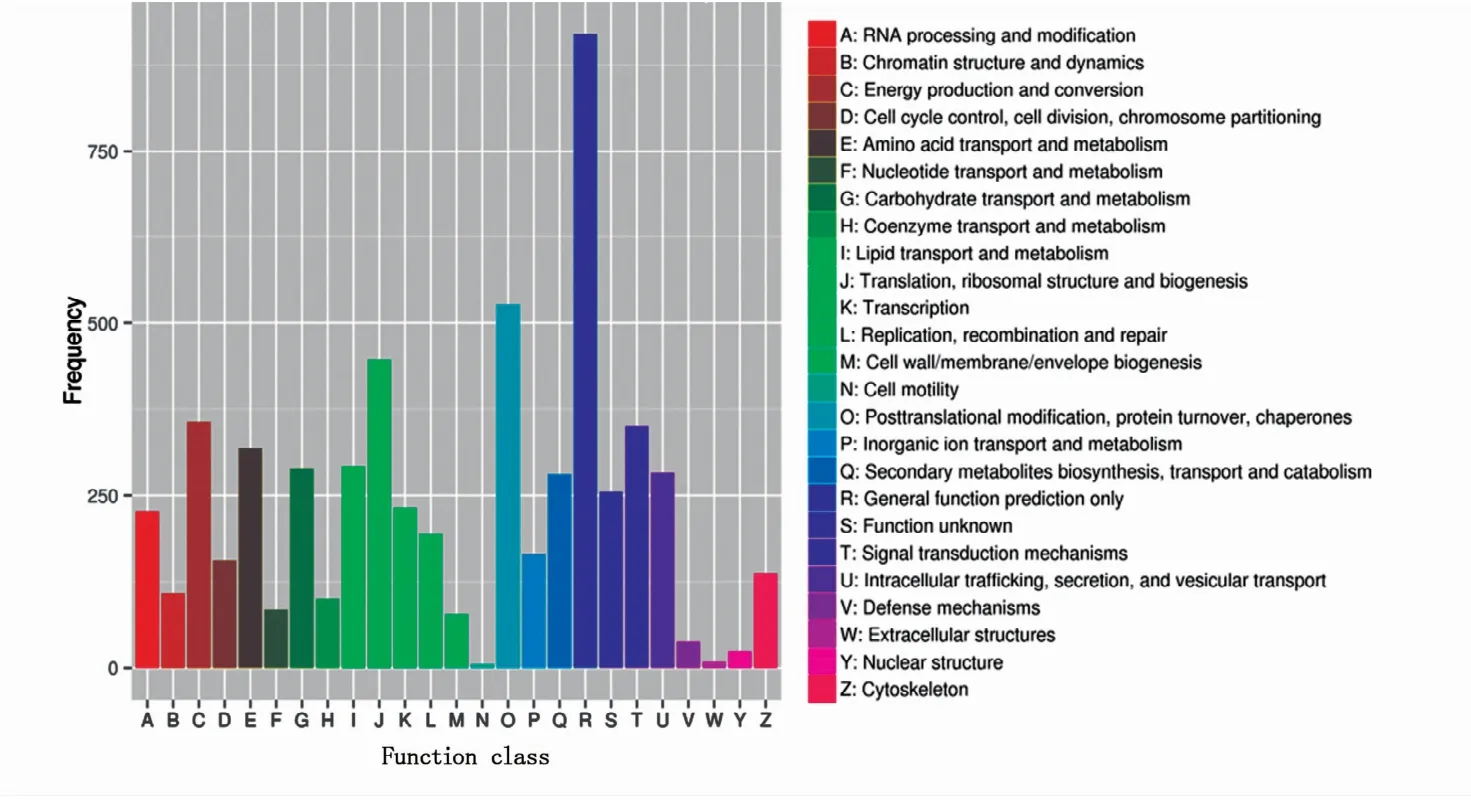
图3 unigene的KOG功能分类
2.3 CDC基因系统进化树分析
系统进化树分析表明(图4),筛选获得的6个CDC相关基因被分为三个亚组。其中CDC3和CDC6在同一亚组,CDC1、CDC5和CDC2在同一亚组,CDC4单独一个亚组。
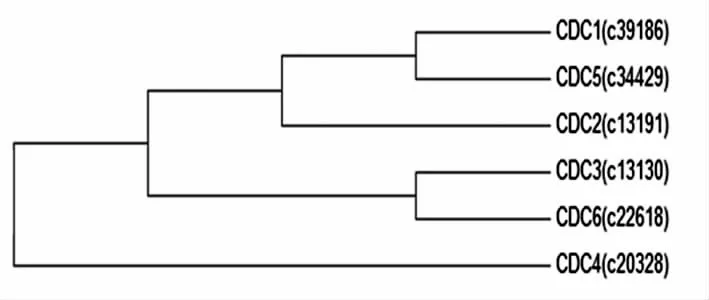
图4 “鲁保一号”菌株CDC基因系统进化树分析
3 讨论与结论
菟丝子能够通过吸器寄生多种作物,严重影响大豆(Glycine max)、牧草和蔬菜的产量和品质[1-3]。“鲁保一号”菌株对菟丝子防效良好,但在经过一段时间的应用后,受制于其致病力退化问题,最终被遗弃[6,7]。由于该菌发现、应用较早,受制于当时的技术条件,并未得到深入的遗传学研究,也没有关于其功能基因研究的报道[3,6,7]。开展该菌株的转录组学研究,初步获得其转录组信息,对于挖据优良基因资源、解决其致病力退化问题等具有重要意义。
转录组测序中获得的unigene片段太短会导致在后续比对注释过程中无法找到匹配序列。本研究采用Illumina Hiseq 2500高通量测序技术首次对“鲁保一号”菌株的转录组进行测序和组装分析,共获得17 031条unigene序列信息,平均长度为1 827.65 bp,能够很好地用于进一步的功能注释和分析,为后续批量分析“鲁保一号”菌株功能基因提供了可能。结合相关生物信息学分析方法,对获得的“鲁保一号”菌株unigene序列信息与各数据库进行比对,进行序列相似性和功能注释分析。NR数据库比对显示,86.04%unigene标注信息与炭疽菌序列一致,这也进一步证明了“鲁保一号”菌株为炭疽菌属;另一方面,13.96%的unigene与其它物种有不同程度的同源性,为“鲁保一号”菌株功能基因的进一步挖掘提供了参考。GO分类进一步显示了“鲁保一号”菌株生长发育过程中基因表达谱的总体情况,其中细胞组分中的细胞成分、细胞器成分,分子功能中的催化活性、结合活性,生物学进程中的代谢进程、细胞进程和单一生物进程涉及的unigene较多,为下一步大量挖掘相关功能基因奠定了基础。基于SSR位点的分子标记在物种遗传图谱构建、遗传多样性分析、相关生物进程功能基因发现和以SSR为分子标记的辅助育种等研究中得到了较为广泛的应用[24-26]。本研究通过查找发现了3 587个SSR位点,接下来可设计并筛选SSR引物,为进一步开发新的SSR标记奠定基础。
前期报道显示,“鲁保一号”菌株连续培养后存在细胞分裂异常现象[7]。本研究以获得的转录组数据库为基础,经过序列比对分析,初步筛选获得了6个CDC相关基因,分属3个亚组。丝状真菌内CDC基因的功能研究表明,不同的CDC基因在生物功能发挥过程中起到不同作用[27];同一亚组内的基因在功能上存在相近或互补的可能,这为下一步集中开展“鲁保一号”菌株CDC基因功能分析提供了参照。实时定量分析表明其中的两个基因在连续培养后表达量显著增加,说明这两个基因可能与连续培养后的细胞分裂异常相关,可能参与了“鲁保一号”菌株致病力退化的调控过程,为下一步的深入研究确立了目标。本研究通过高通量测序获得了“鲁保一号”菌株的大量转录组信息,为其基因克隆、分子标记发掘和基因组学研究等提供了有价值的数据。
猜你喜欢
中国人兽共患病学报(2022年9期)2022-10-19
卫星应用(2022年3期)2022-05-23
中国典型病例大全(2022年11期)2022-05-13
中国典型病例大全(2022年11期)2022-05-13
中国新通信(2022年4期)2022-04-23
草地学报(2022年3期)2022-03-28
今日农业(2021年11期)2021-11-27
科学导报(2021年29期)2021-06-03
中国生殖健康(2020年4期)2021-01-18
科海故事博览·下旬刊(2019年6期)2019-04-16
