
过渡金属磷化物的制备及电催化析氢性能提升策略
2021-05-17 08:54季小好王祖民陈晓煜于然波
高等学校化学学报 2021年5期
季小好,王祖民,陈晓煜,于然波
(1.北京科技大学冶金与生态工程学院,北京100083;2.中国科学院过程工程研究所,北京100190)
氢能作为一种清洁能源来源广泛,为宇宙中最丰富的元素(占所有原子的90%以上),其燃烧热值为液化石油气的2.5 倍,汽油的3 倍,且无毒,燃烧后无有害产物. 作为一种可持续能源,氢能在弥补能源短缺、环境保护方面具备强大优势. 目前最常用的制氢方法主要有电解水制氢、化石燃料重整制氢、生物质热转化和发酵制氢及光催化裂解水制氢等.
自William Nicolson 和Anthony Carlise 于1800年发现电解水产生氢气之后[1],人们逐渐发现电解水制取的氢气纯度高,不需要复杂的提纯步骤,随后便意识到该方法在实际应用层面有着极大的发展潜力和优势,目前最成熟且已大批量商业化规模使用的是碱性电解水制氢技术. 水电解的化学反应为:2H2O→O2+2H2,整个反应由两个电化学半反应组成[2],分别是阳极的析氧反应(OER)和阴极的析氢反应(HER)[3]. 水分解的实际施加电势(E)可表示为[4,5]:E=E0+iR+η,电解水理论分解电压E0为1.23 V[6]. 在实际电解过程中,存在着内阻(iR)与过电势(η),导致电解电压远大于1.23 V,而高效电催化剂的引入可以明显降低反应过电势η和加速反应动力学,提高电解效率. 对于析氢反应,已经有许多性能优异的贵金属催化剂,然而在实际应用中仍存在许多亟待解决的问题,如高温下的电极材料与电解池的腐蚀问题[7]. 此外,更为现实的问题是贵金属的储量有限. 在此背景下,非贵金属催化剂成为研究热点. 目前的研究主要为控制和优化电催化剂的电子结构和催化剂本身的微观结构,从而提升催化剂的内在活性达到改善电极材料电催化性能的目的[8,9]. 各类金属的析氢内在活性通常使用氢吸附自由能与交换电流来评估. 根据Sabatier原理[10],当催化剂对质子吸附能力较弱时,二者难以结合不能发生有效的催化析氢反应;相反,如果催化剂对质子吸附能力过强,结合的氢原子从催化剂表面脱附困难,从而导致催化剂表面的活性位点被永久占据,致使催化剂中毒. 对于反应:,从物理化学的角度来看,在析氢过程中时刻伴随着金属-氢键的生成和断裂,所以可以通过计算析氢反应过程中的氢吸附自由能ΔGH*评估H*吸附和H2脱附[11],进而判断催化活性[12]. Sabatier原理规定,在理想条件下ΔGH*应该为零且析氢反应的交换电流i0最高[10]. Parsons建立了一个“火山类型”图,将i0值与ΔGH*相关联[13],有关公式如下:
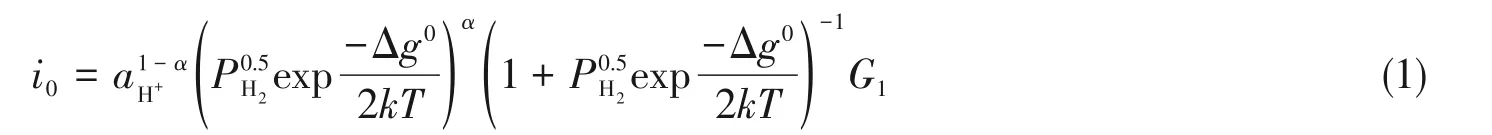
式中:i0(A/cm2)为交换电流密度;aH+为溶液中质子浓度;Δg0(kJ/mol)为反应标准吉布斯自由能,即为ΔGH*;PH2为氢气分压;k(kJ·mol-1·K-1)为吸附平衡常数;T(K)为反应温度;α为传递系数;G1(A/cm2)为实验测得量,根据式(1),绘制出i0与ΔGH*的关系图[14](图1).
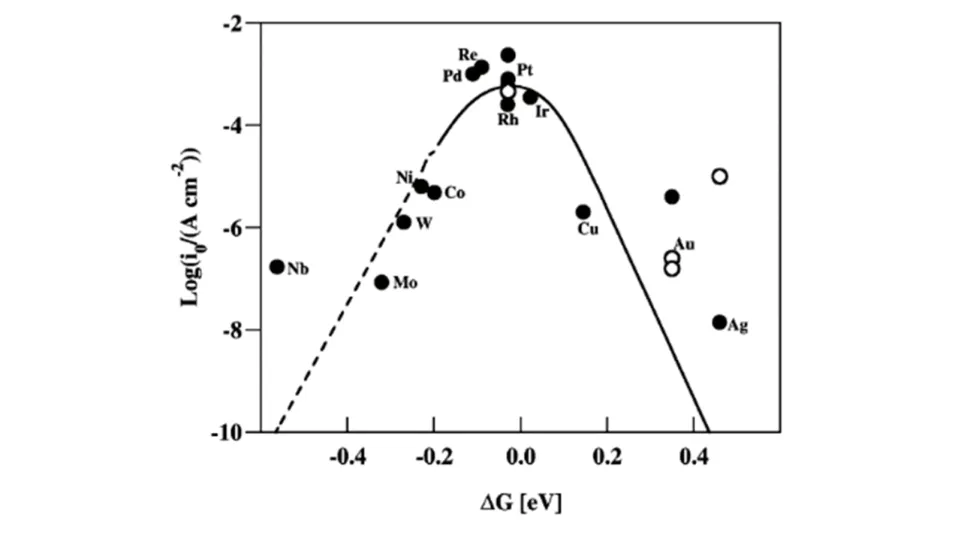
Fig.1 HER volcano plot calculated by theory[14]
得益于理论的发展,催化剂的研发由早期的“试错”过渡到现在的理论与实验相结合的阶段,伴随着计算科学的发展,通过密度泛函理论(DFT)预估氢吸附自由能ΔGH*与实验测得的交换电流i0可以绘制出更为详细的火山图[15].由于非贵金属来源广泛,价格低廉,人们便将目光放在火山图两边的非贵金属上,特别是过渡金属基催化剂. 如过渡金属硫族化合物(Transition metal dichalcogenides,TMDs)、过渡金属氧化物(Transition metal oxides,TMOs)、过渡金属碳化物(Transition metal carbides,TMCs)、过渡金属氮化物(Transition metal nitrides,TMNs)以及过渡金属磷化物(Transition metal phosphides,TMPs)等. 在电催化析氢中,层状TMDs 在酸性条件下被公认为有效的HER 催化剂,而其在中性和碱性中催化活性较低[16],其催化活性可能比酸性溶液低2~3 个数量级[17].另一方面,TMDs 的半导体性质和其活性位点分布在结构边缘的特点无疑会对电催化带来不利影响.所以,对TMDs来说,通常需要将合成的TMDs剥落[18],或使用等离子体与氢气下退火形成缺陷[19],来改善其电导率并增加活性边缘数量. 但是这些处理过程复杂且不经济. TMOs电催化剂与其它种类的过渡金属催化剂相比,其电导率相对较低,大多数TMOs是半导体[20],固有活性不高,且在应用过程中耐酸碱腐蚀性较差. 另外TMCs和TMNs也用作HER领域中的电催化剂. 但是它们的合成涉及高温处理或氨还原后再退火[21,22],导致合成过程复杂,成本较高.
相比上述其它过渡金属化合物的一些缺点,TMPs具有近似球形的三角棱柱单元结构和金属导体性质,而TMPs的几何结构能使更多的晶体表面暴露出来,自然为电催化提供了更多的活性位点[23],并且大多数表现出较好的电催化导电性等优点. 此外TMPs在pH=0~14的介质中均具有高活性,高稳定性和近100%的法拉第效率,因此TMPs具有较高的研究价值和应用前景. 常见析氢过渡金属化合物的简略对比列于表1.
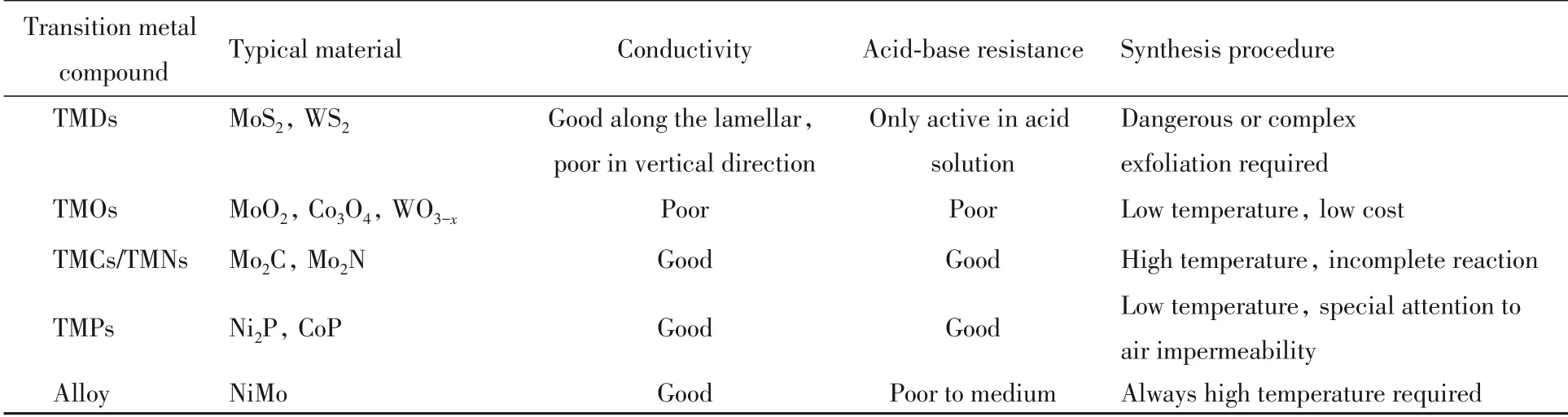
Table 1 Comparison of common hydrogen evolution transition metal compounds
1 过渡金属磷化物简介
TMPs 的发现可以追溯到18 世纪,近两百年来TMPs 没有得到广泛的应用. 直到20 世纪60 年代,TMPs 才逐渐被应用于加氢脱硫(Hydrodesulfurization,HDS)、加氢脱氮(Hydrodenitrogenation,HDN)、加氢处理(Hydroprocessing,HPC)、农药、光催化降解、锂离子电池等领域,最近则成为了热门的HER催化剂[24]. 20世纪90年代,Paseka[25,26]和Burchardt[27]的开创性研究表明,非晶态过渡金属(如Ni,Co和Fe)磷化物在碱性条件相对较高的过电势下具有较好的HER活性. 但是当时人们认为,这些电极的高活性来源于电化学过程中通过氢吸附调整的金属电子结构,而不是催化剂本身. 2005 年,Liu 和Rodriguez[28]通过理论计算预测Ni2P的(001)晶面可能是对HER的极佳催化表面,甚至优于基准Pt基催化剂. 直到2013 年,Popczun 等[29]从实验验证了Ni2P的(001)晶面优异的析氢活性,从此TMPs正式进入了电催化析氢领域. 此后,不同种类的纳米结构TMPs相继出现,如Ni2P,CoP[30],MoP[31],FeP[32]等.
1.1 过渡金属磷化物的结构与性质
通常,元素周期表中的几乎任何一个金属元素都可以与磷元素形成一种或数种磷化物,且物相复杂,结构多变. 其根本原因在于磷元素与金属元素的结合方式,由于磷原子的半径(0.109 nm)较大,在结合中磷原子不能进入金属原子密堆八面体中,金属原子会形成三棱柱并围绕着磷原子. 由于磷的掺入,使得原本金属原子之间的距离增加,导致金属原子之间的相互作用力减弱,d带收缩[33],使得费米能级处态密度增加,表现类似于常见的贵金属元素. 磷元素与过渡金属结合形成TMPs时,不仅会形成金属-磷键(M-P),磷-磷键(P-P),也会形成金属-金属键(M-M),因此会有多种化学计量比的过渡金属磷化物[34]. 常用来合成过渡金属磷化物的金属元素有Ni,Co,Mo,W,Fe 和Cu 等,它们的常见结构如Scheme 1所示.
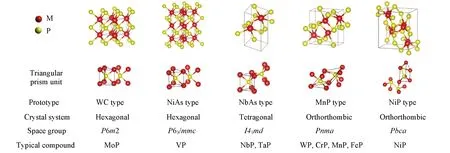
Scheme 1 Common structures of transition metal phosphide
在过渡金属磷化物中,金属种类与磷含量不同形成的结构往往不同,其性质千差万别. 如Ni2P,CoP,MoP,FeP 等材料拥有极好的稳定性和催化性,在电解水制氢中得到广泛的应用;Cu3P具有较大的比容量,常被用作电池负极材料;IrP 材料硬度大,化学性质稳定;NbP 具有明显的巨磁电阻效应.已知过渡金属磷化物中磷元素与金属元素可具有不同的化学计量比,按照磷原子(P)与金属原子(M)个数之比可分为富金属磷化物(M/P ≥1)和富磷磷化物(M/P <1)两类. 相比富金属磷化物,富磷磷化物的稳定性较差,其富含非金属元素磷,因而导电性较纯金属差,多数属于半导体. 而富金属磷化物具有良好的导热及导电能力. 一般来说在电催化析氢领域常用富金属磷化物作为电催化剂.
1.2 过渡金属磷化物的合成
在20世纪50~60年代,金属磷化物的制备条件较为苛刻,通常使用安瓿技术或电弧熔炼技术合成金属磷化物[35],在合成过程中常使用易燃的磷和高毒性的膦作为磷化试剂,且反应大多需要高温高压,这对大规模的工业合成和催化应用不利[36]. 随着合成化学和纳米科学的快速发展,一些新颖的合成方法相继出现. 根据合成体系将其划分为固相合成、气相合成与液相合成3大类.
1.2.1 固相合成法 固相合成法通常使用的是固体前驱物,在高温下直接反应,是一种普适的制备金属磷化物的方法,一般在充满保护气的管式炉或在真空环境中进行,利用固体前体与气体小分子(由固体分解、升华等过程气化生成)反应. 其中固体前体为纳米金属颗粒、纳米金属氧化物和纳米金属氢氧化物等,而气体小分子常为PH3或磷蒸气.
最常见的合成步骤为将单质红磷和相应金属粉末按照目标产物的化学式进行摩尔配比,在真空或惰气保护下密封在石英管中高温反应,该过程可以表示为:M+P→MPx. 如Huang等[37]利用等离子蒸发法制备出Mo颗粒,然后将其与红磷放置于密闭石英管中在不同计量比下成功合成了颗粒状的MoP与片状的MoP2,颗粒状的MoP粒径大小为20 nm左右[见图2(A)~(C)]. 由于这种高温反应会产生高活性的磷蒸气,因此对实验设备要求较高,需要保证制备过程中整个设备的密闭性与尾气处理的安全性. 也有使用磷的氧化物与金属氧化物在氢气下共还原来制备过渡金属磷化物. 如Yao等[38]使用合成的MOs(NiO,CoO,MoO3)与P2O5在氢气气氛下合成了Ni2P,Co2P 和MoP[见图2(D)~(G)]. 但是使用上述固相合成法通常需要高温高压(通常的反应温度高于600 ℃[39]),且制备出的金属磷化物尺寸较大.
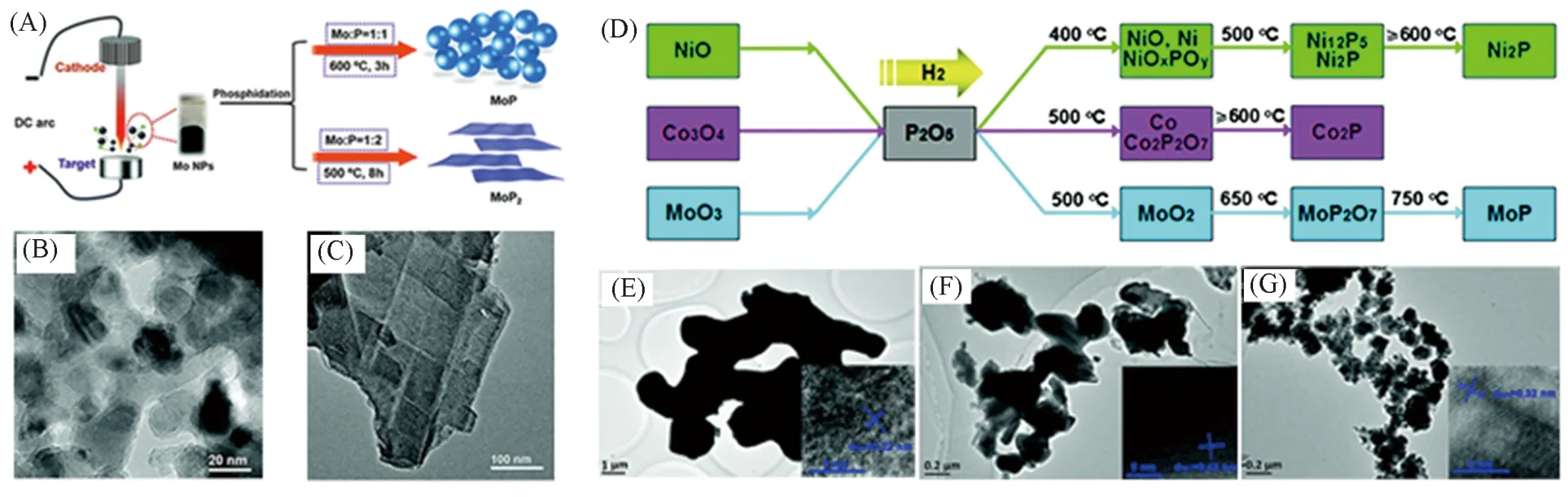
Fig.2 Schematic diagram of the preparation process of MoP nanoparticles and MoP2 nanosheets(A),TEM images of MoP nanoparticles(B)and MoP2 nanosheets(C)[37],schematic diagram of the synthesis of Ni, Co, Mo oxide and P2O5 co-reduction to prepare the corresponding phosphide(D),TEM images of S-Ni2P(E),S-Co2P(F)and S-MoP(G)[38]
为了降低制备过程中的温度,通常选择PH3作为磷化气体,而PH3通常由NaH2PO2在低温下分解得到,而金属前驱体多为金属氧化物与金属氢氧化物. 具体做法为:将NaH2PO2和金属氧化物前体分别放置在两个瓷舟中,前者在上游,后者在下游,温度超过250 ℃时,NaH2PO2迅速分解,同时释放出PH3,PH3可以进一步与金属氧化物前体反应生成金属磷化物. 反应可以表示为:2NaH2PO2→Na2HPO4+PH3↑;PH3+MyOz→MPx+nH2O或PH3+My(OH)z→MPx+nH2O. 此方法经Sun等[40~42]发展后已成为一种磷化的常用方法. 该反应路线温度(300 ℃左右)较低,且磷化时间(2 h左右)较短,可以最大程度地保留金属氧化物或金属氢氧化物前体的形态. 如Xu等[43]在碳布上首先水热生长纳米针状分支Co(OH)F,然后使用钼酸钠为钼源水热掺杂钼元素获得Mo-Co(OH)F,最后使用NaH2PO2热分解产生的PH3磷化最终得到Mo-CoP 纳米片,Mo-CoP 纳米片基本保持上一步水热生长的Co(OH)F 形貌[图3(A)~(C)].
1.2.2 液相合成法 液相合成法是目前研究最为充分的方法. 该方法对产物的大小、形状、组成及结构都可以进行灵活操控. TMPs的液相合成常采用溶剂热法. 通常溶剂为1-十八烷基、辛基醚和角鲨烷等高沸点溶剂,使用有机磷作为磷源,包括三辛基膦(Tri-n-octylphosphine,TOP)、三苯基膦(Triphenylphosphine,TPP)、三辛基氧化膦(Tri-n-octylphosphine oxide,TOPO)及其类似物. Wang等[44]使用TOP作为有机配体,乙酰丙酮镍作为金属前体,在溶液中通过调控金属源与磷源的配比,在不同的温度条件下分别得到了NixPy空心纳米颗粒,NixPy非晶纳米颗粒和NixPy固体结晶纳米颗粒[图4(A)~(G)]. 反应温度在220 ℃以上时,TOP分子中的C-P键断裂,随后磷原子可以与金属原子配位最后生成金属磷化物[29]. 通过控制反应温度在240 ℃得到了非晶态的NixPy纳米颗粒,当继续加热至300 ℃时转变为晶态的NixPy纳米颗粒,当原料比例改变后在300 ℃时得到了中空NixPy纳米颗粒. 此方法要求严格的惰性气氛条件,以防止释放出高度易燃的磷化氢.
1.2.3 气相合成法 气相合成法常用的反应物为含膦小分子气体(PH3,PMe3,PEt3等)和金属有机单源分子前驱物([Co(CO)3PEt3]2,Fe(CO)4[PPh2CH2CH2Si(OMe)3]等). 常用的气相合成法为化学气相沉积法,Schipper 等[45]使用单源分子前驱体化学气相沉积法成功制备了一系列铁基磷化物[图4(H)~(K)]. 此方法所需仪器较为复杂,存在前驱物不易合成、不稳定的缺点,且制备出的TMPs常为附着在基底上的涂层.
1.2.4 其它合成方法 除上述方法外,为了使合成过程更加安全、快捷,一些新颖的策略也在不断涌现.(1)电沉积法[46,47],与有机溶液合成相比,电化学沉积要温和得多[48],电沉积过程主要在室温下进行,并且不会释放出高毒性物质. 由于金属离子Mn+和H2PO2-还原反应,金属磷化物可以轻易地沉积到导电电极表面. 反应式如下:,不过其生成的金属磷化物具有非晶态特征和复杂的组成. 目前已有报道采用电沉积方法制备的TMPs 为Co 和Ni 的磷化物[46,47].(2)可以通过金属有机骨架(MOF)衍生的途径来合成纳米结构TMPs/碳复合材料. 此方法合成的纳米结构有较高的比表面积、丰富的孔结构、超细的尺寸和优良的导电性[49,50].(3)为了缩短合成时间,最近开发了一种微波辅助制造纳米结构TMPs的策略[51,52]. Zhang等[52]以离子液体为媒介,四丁基氯化膦为磷源,使用微波辐射仅2 min 便合成了不同形貌的Ni2P 与Ni12P5.(4)与不安全的无机磷源相比,环保、无毒的生物质磷源更具吸引力[53]. 如Zhang等[54]用植酸作为磷源合成TMPs的新颖策略. 由于植酸的6个磷酸基团与金属阳离子之间的强配位作用,其很容易与金属离子形成植酸-金属的交联结构. 植酸-金属的交联前体热解即得到金属磷化物. 该方法具有大规模合成的潜在优势.
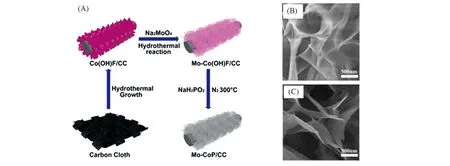
Fig.3 Schematic diagram of the synthesis of Mo-CoP nanosheets(A), SEM images of Mo-Co(OH)F(B)and Mo-CoP(C)[43]
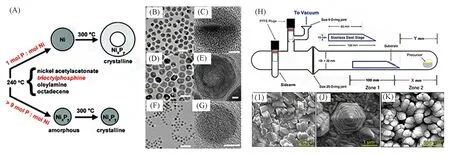
Fig.4 Schematic diagram of NixPy nanoparticle synthesis(A),TEM images of crystalline NPs of nickel NPs(B,C), hollow NixPy NPs(D,E), solid NixPy NPs(F,G)[44], the deposition apparatus(H), SEM images of FeP(I),Fe2P(J),and Fe3P(K)on FTO[45]
上述根据合成体系将其划分为固相合成、气相合成与液相合成3 大类,最后还介绍了一些新方法. 不难发现,固相法合成温度较高,气相法的前驱物合成复杂不稳定,液相法需使用较为危险的有机溶剂,且3种方法在制备过渡金属磷化物的过程中均会放出有毒尾气,需要惰性气氛和严格的密封条件. 因此需要寻找更为安全无毒的磷源和较为缓和的反应条件. 目前,电化学和生物质合成的策略较为符合这一要求,但仍需进一步探究. 表2总结对比了不同方法的优缺点,表3~表5则总结了不同制备方法下各种过渡金属磷化物的合成条件与析氢性能[30,32,41,55~75].
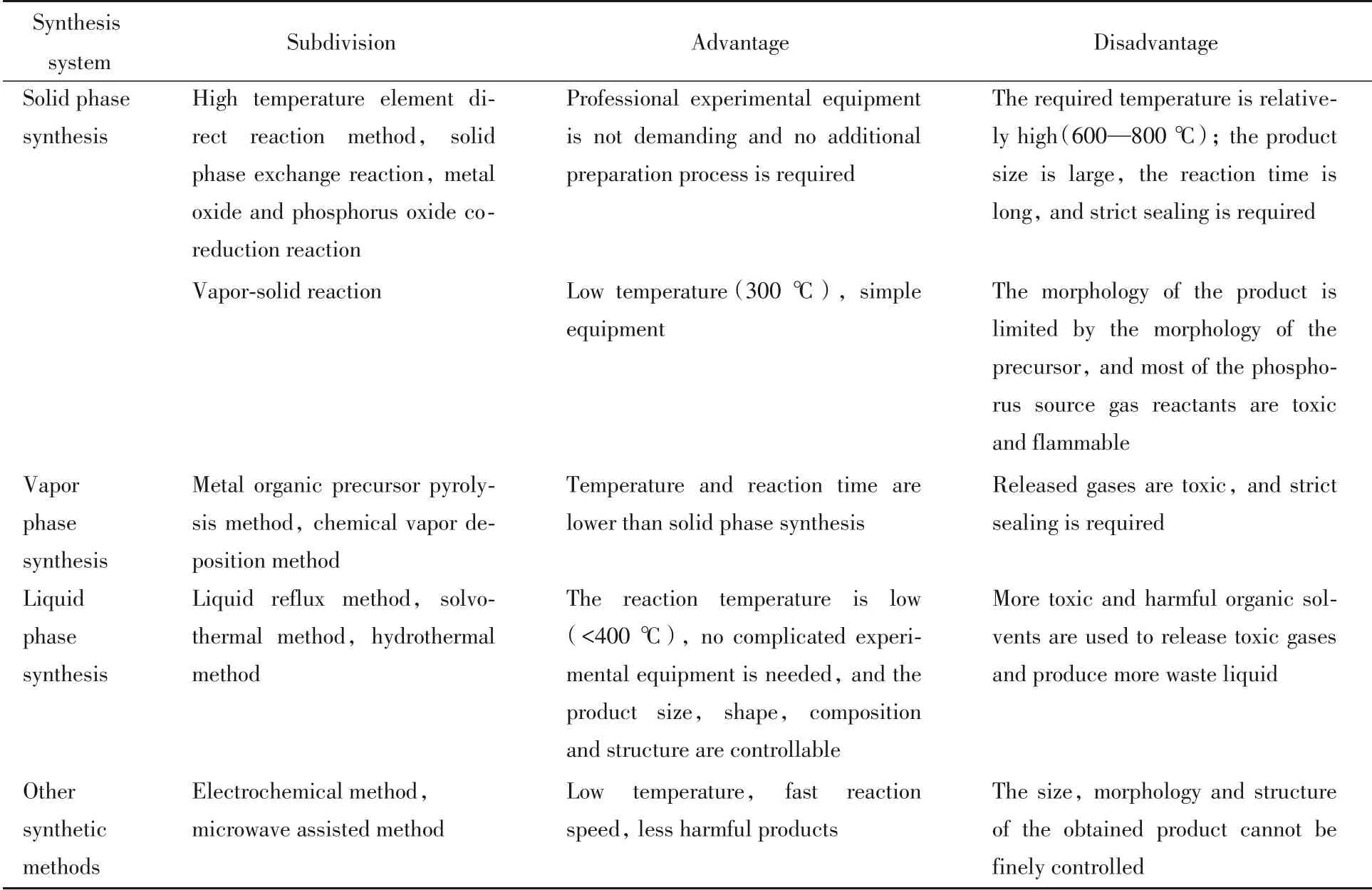
Table 2 Advantages and disadvantages of common synthesis methods for transition metal phosphides
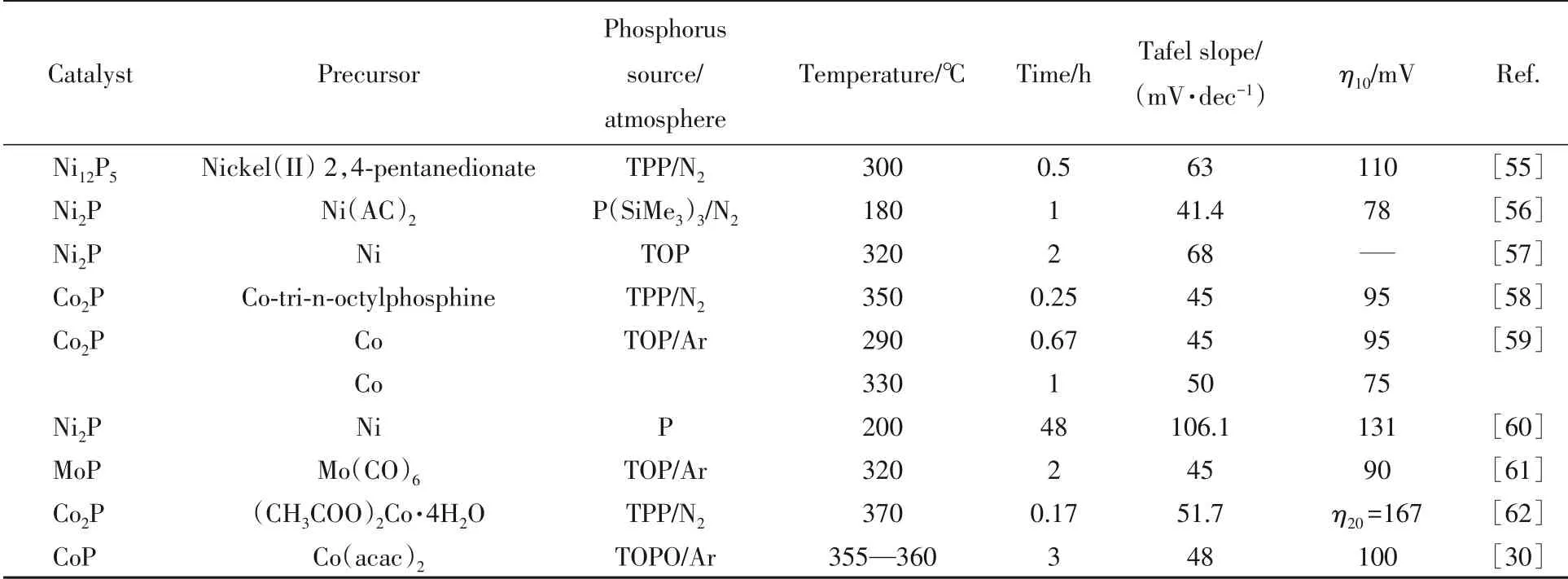
Table 3 Synthesis conditions and hydrogen evolution performance in 0.5 mol/L H2SO4 of various transition metal phosphides prepared by liquid phase method

Table 4 Summary of the synthesis conditions and hydrogen evolution properties of various transition metal phosphides prepared by electrochemical methods at room temperature
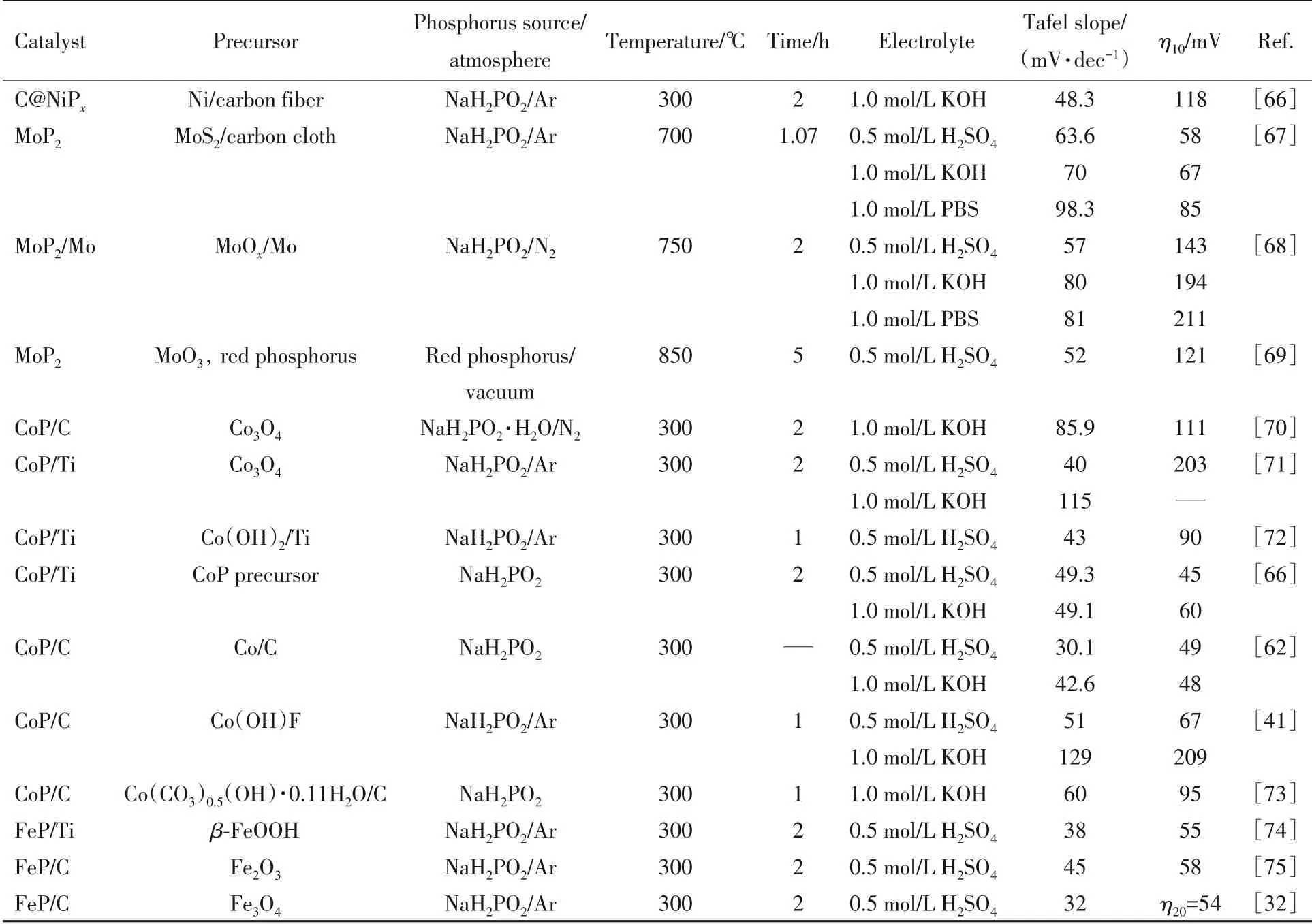
Table 5 Synthesis conditions and hydrogen evolution performance of various transition metal phosphides prepared by gas-solid method
2 过渡金属磷化物的析氢反应机理
2.1 过渡金属磷化物中金属元素对电催化析氢性能的影响
结合理论研究与实验验证,TMPs具有优良的析氢催化活性. 在析氢催化中,过渡金属磷化物中金属元素不同,使得其理论计算所得的氢吸附自由能不同. Jaramillo 等[76]利用密度泛函理论计算来预测一系列铁、钴、镍、钼基混合金属过渡金属磷化物与单金属磷化物的氢吸附自由能ΔGH,并将这些结果与实验确定的HER活性进行比较,从而确定了常见的过渡金属磷化物中最佳的金属磷化物催化剂(图5).
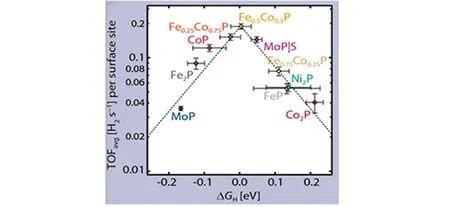
Fig.5 Activity volcano for the HER showing the average TOF at η=100 mV as a function of ΔGH[76]
在上述过渡金属基磷化物系列中,发现Fe0.5Co0.5P是最活跃的析氢反应催化剂,具有接近零的ΔGH理论值(0.004 eV)和(0.19±0.01)H2s-1的转换频率(TOF)实验平均值. 此外,该研究还指出,单金属CoP具有与Fe0.5Co0.5P可比的HER活性,其理论ΔGH=-0.09 eV,不过实验得到的TOF平均值约为0.18 H2s-1,与Fe0.5Co0.5P相近,表明仅改变金属元素掺杂量虽然其理论ΔGH明显改善,但实际的产氢量并不一定有显著提升. 这是因为H结合位点存在多种可能,即可以吸附到金属位点或磷位点,也可吸附在金属与磷之间的桥位点或其它类型的位点上. 此外,TMPs表面上H的吸附通常伴随着表面结构的动态重排过程[76]. 因此通过改变或掺杂金属元素时,应综合分析其各种因素对最终催化结果的影响,最好将理论计算与实际实验相结合来评判不同金属元素与性能之间的相互关系.
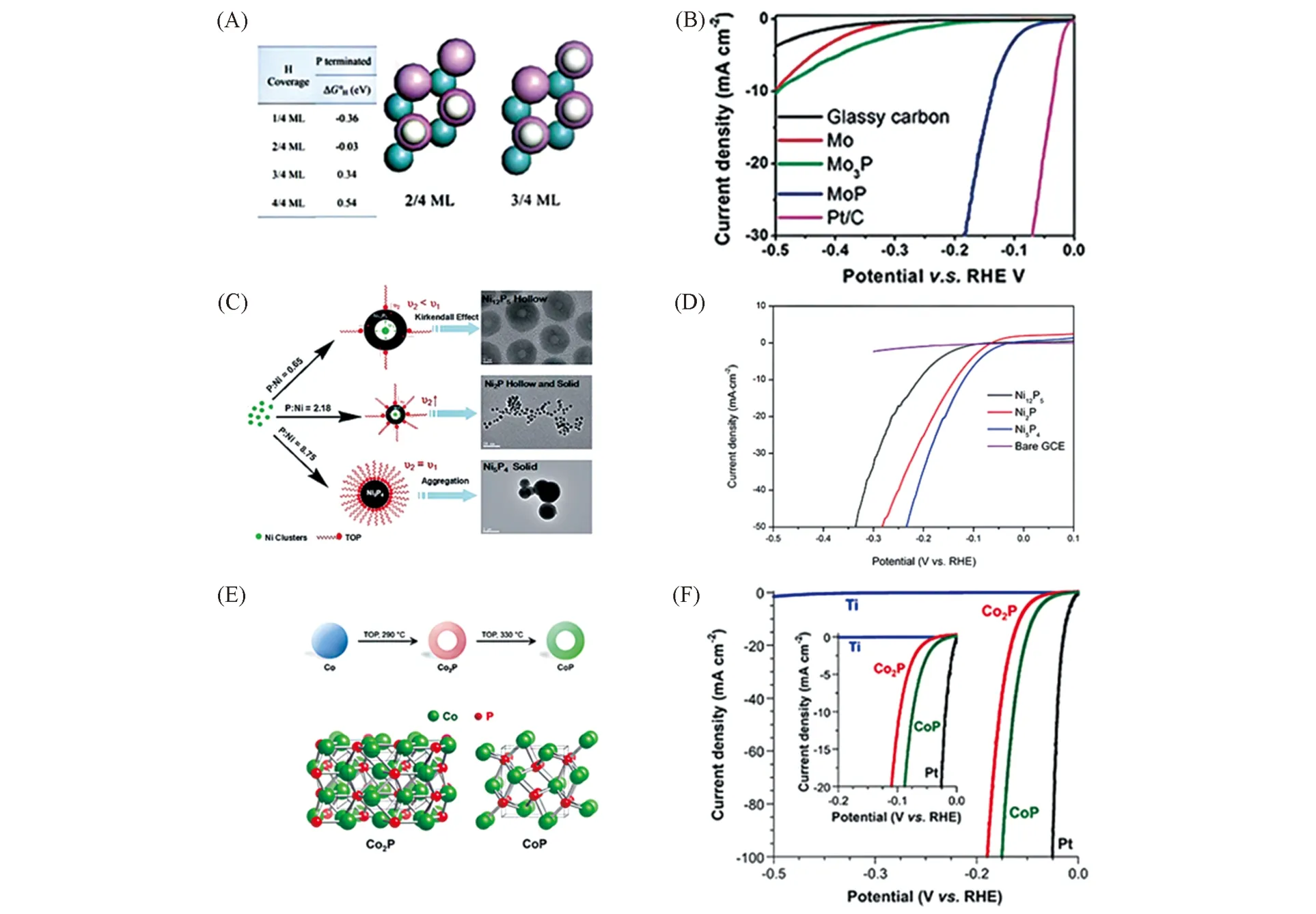
Fig.6 Gibbs free energy of H adsorption on Mo(110) and MoP(001)(A), polarization curves of MoP(B)[77], possible formation mechanism of the as-synthesized nickel phosphide NCs(C), LSV curves of bare Ni5P4 NCs(D)[78],schematic showing the multistep reaction pathway for Co2P and CoP and crystal structures(E),polarization data for Co2P/Ti and CoP/Ti(F)[59]
2.2 磷元素在过渡金属磷化物电催化析氢中的作用
磷在TMPs中的晶格位置可以看作是将磷原子掺杂到过渡金属的晶格中,同一种金属对应的磷化物中磷原子可以有不同的含量,形成不同计量比的TMPs,如CoP 和Co2P,CuP 和Cu3P,FeP 和Fe2P,Ni2P,Ni5P4和Ni12P5等.
之前认为,富磷TMPs的性能优于富金属TMPs. Wang等[77]通过DFT计算钼的磷化物,发现P原子对TMPs的电催化析氢性能起着至关重要的作用. 由于P原子的电负性强于Mo原子,P原子与Mo原子的结合会使电子云向P原子偏移,通过DFT计算发现,在电化学反应中,当H覆盖率在2/4单分子层到3/4单分子层时,带负电荷的P原子吉布斯自由能接近零,更易与带正电的H+结合,这便成为电催化析氢性能位点. 其HER 性能规律为MoP>Mo3P[图6(A)和(B)]. Liu 等[78]利用液相法制备了Ni5P4,Ni2P和Ni12P5单分散的纳米晶体. 在0.5 mol/L H2SO4中,经过实验测试其析氢性能时,发现其性能规律为Ni5P4>Ni2P>Ni12P5[图6(C)和(D)]. 这种优异的催化活性归因于磷含量较高的Ni5P4NCs 中Ni原子与具有较高负电荷P原子之间较强的组合效应(Assembly effect). 之后,Schaak等[59]关于过渡金属Co的磷化物研究也发现了类似的性能规律,他们比较了形态类似的Co2P和CoP纳米晶体的析氢反应活性,发现析氢活性大小规律为CoP>Co2P,即富磷相大于富金属相[图6(E)和(F)].
已知碱性介质中的析氢机理与酸性介质中的不同,上述3例的HER测试均在0.5 mol/L H2SO4中进行,表明富磷相的析氢活性大于富金属相,这一经验规律在碱液中是否适用尚未确定. 最近,Kim等[34]通过热解次磷酸钠低温磷化石墨片上磷化Ni(OH)2纳米阵列得到了Ni2P,Ni5P4和NiP2纳米片阵列,在1 mol/L KOH 的碱性溶液中测试,虽然催化剂表面出现氢氧化物与磷酸盐,但是HER性能大小顺序仍满足NiP2>Ni5P4>Ni2P的规律[图7(A)和(B)].
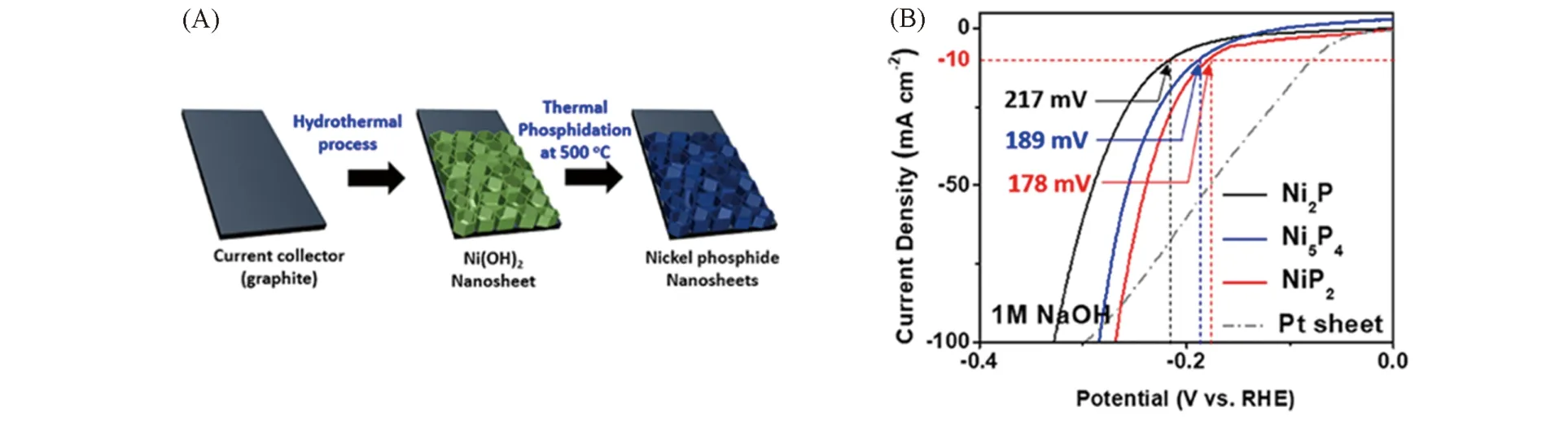
Fig.7 Schematic for the preparation of nickel phosphides(A), the polarization curves of nickel phosphide nanosheets with different phases in 1 mol/L NaOH(B)[34]
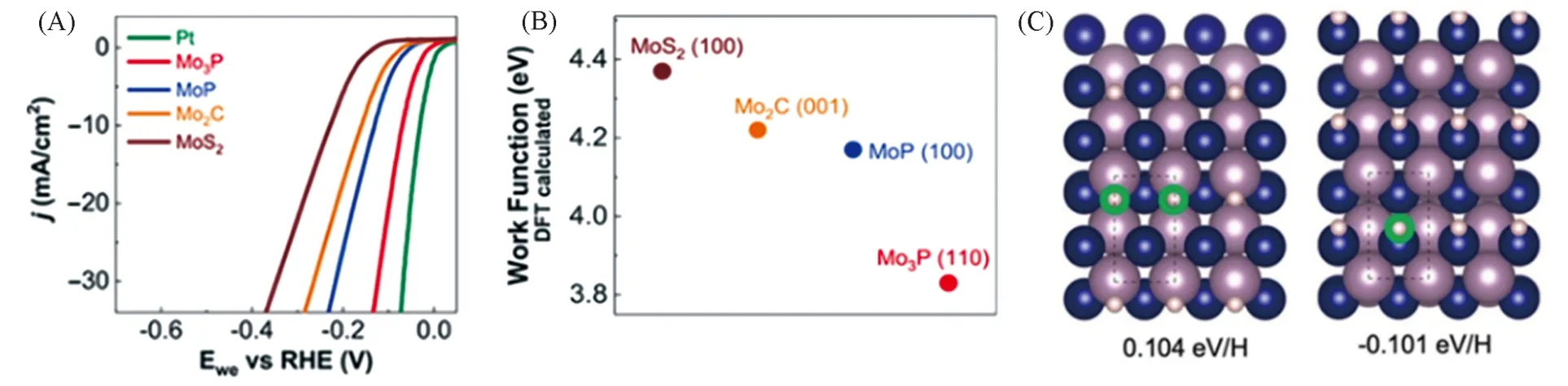
Fig.8 Polarization data in 0.5 mol/L H2SO4 for Mo3P, MoP, Mo2C and MoS2(A), comparison of absolute work function for each catalyst(B), top view of hydrogen adsorption on MoP(100) corresponded to two different adsorption sites of Mo sites and P sites(large purple atom:Mo;blue atom:P;small purple atom:H)(C)[15]
Wang等[77],Liu等[78],Schaak等[59]与Kim等[34]的研究表明,对应于某种金属组成的金属磷化物,提高P 的含量都有效地提高了该类金属磷化物的电催化析氢性能,即富磷TMPs 的性能优于富金属TMPs,与早期认识相符. 然而,Schipper等[45]对各种化学计量比的铁基磷化物薄膜研究却发现HER活性遵循Fe3P>Fe2P>FeP的趋势,即P含量较低的铁基磷化物的析氢性能较高. 其原因是在氢吸附自由能为零时,富Fe 磷化物(Fe3P 和Fe2P)表面具有较高的氢覆盖率. 同样地,最近Asadi 等[15]结合实验和理论计算发现,低磷的Mo3P纳米粒子比富磷的MoP具有更高的HER活性. 其中Mo3P为四方晶系,每个Mo原子与2个或4个P原子配位,而MoP结晶为六方晶系,Mo则由P原子形成六配位. 两者的结构有着很大的不同. Mo3P纳米薄片显示出很高的电催化HER行为,在10 mA/cm2的电流密度下仅需69 mV的过电势,小于对应电流密度下MoP 的过电势(130 mV)[图8(A)]. 因Mo3P 具有较低的功函数[图8(B)],其电荷转移较MoP容易. 析氢反应模拟计算揭示,对于Mo3P而言,氢倾向于吸附在Mo-Mo桥位而不是P位点,而MoP与Mo3P相反,氢倾向于吸附在P位点而不是Mo位点. 显然,两者的活性位点不同,如图8(C)所示.
值得注意的是,早期观点所认为的富磷TMPs 的性能优于富金属TMPs 的工作中,如Wang 等[77]制备的MoP与Liu等[78]制备的Ni5P4的活性位点均为P位,而后期的一些“反例”中,如Schipper等[45]制备的Fe3P与Asadi等[15]制备的Mo3P的活性位点为M-M金属桥位. 所以单纯的通过P含量来预测过渡金属磷化物的析氢反应活性变化并不完全正确,应考虑P含量变化后活性位点的改变与氢吸附之后的表面结构重排等因素的影响[76]. 而前期认为富磷较优,可能是因为其类似于氢化酶结构,而这个结构的活性位点大多是M-P桥位,所以富磷相性能较优. 然而磷含量变化会导致活性位点的改变,当其活性位点是P位或M-P桥位时,富磷相性能较优,但当活性位点在金属M位或M-M金属桥位时富金属相较优.
2.3 过渡金属磷化物电催化析氢反应机制
首先,根据电解水产氢机理理论,电解水中的HER反应为发生在阴极电极表面的多单元步骤:首先是电化学氢吸附步骤(Volmer 反应),在此步骤中阴极上的活性位点会吸附质子,随后吸附的质子会经过两种不同的路径产生H2,分别为电化学脱附步骤(Heyrovsky 反应)和复合脱附步骤(Tafel 反应).因为过渡金属磷化物析氢反应机理类似,因针对Ni2P的研究颇多,以Ni2P为例简单介绍一下其析氢反应机制.
2005年,受[NiFe]氢化酶的HER催化活性的启发,Liu和Rodriguez[28]预测Ni2P的(001)晶面可能是对HER 的极佳催化表面,甚至优于基准Pt 基催化剂. 根据他们的DFT 计算,由于P 原子的电负性更强,电子可以部分地从金属原子转移到相邻的P原子,产生部分带负电的P(δ-)位点作为质子受体和部分带正电的金属M(δ+)作为氢化物受体. 这种吸附不同于纯金属表面,Ni2P的(001)表面上暴露的质子受体和氢化物受体同时存在,氢强烈地束缚在Ni的中空点位,但是在P的协助下吸附状态的氢可以很容易地从Ni2P的(001)面脱附下来,也就是说TMPs中的金属和磷协同促进了HER过程.
2015 年,Laursen 等[79]进一步详细解释了酸性条件下Ni2P 的(001)晶面上析氢反应机制. 酸中的HER 机理可以分3 个步骤进行.(1)电子转移到结合到Ni3三角位点吸附的质子上;(2)电子转移给Ni-P键上吸附的质子,这一步被认为是限速步骤;(3)形成H2. 通过速率步骤可以看出,Ni-P键的长度可能是一个合理的反应描述符,随着键长增加,P上的电子局域性会增强,从而降低质子结合的障碍(图9).
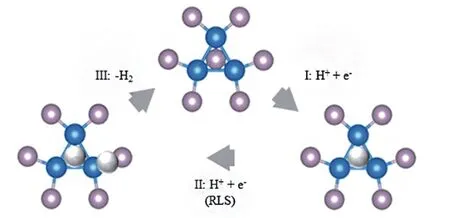
Fig.9 Schematic representation of the reaction mechanism suggested by Liu and Rodriguez[28](blue:Ni;purple:P;white:H)[79]
3 提升TMPs析氢性能的常用策略
尽管纯相TMPs表现出优异的电催化析氢活性,但其中吸附能的大小与催化剂的电导率仍与贵金属存在着差距. 若想提升TMPs的析氢性能,必须对催化反应过程有一定的了解. 在催化过程中,催化剂表面活性位点上时刻存在着反应物的吸附、旧键断裂、新键生成、电荷的转移及反应产物的脱附等过程[80]. 因此,催化剂表面活性位点的本征活性和其与周围溶液环境之间的传质均会对催化反应产生重大影响[81]. 从催化剂设计的角度出发:(1)提升催化活性位点的本征活性;(2)增加催化活性位点的数量. 提升本征催化活性一般从电子结构和能量角度对催化剂进行调整和设计,常用元素掺杂与界面调节来提升活性位点的固有催化活性. 而基于几何因素,对催化剂进行结构上的构筑通常可以改善催化剂与周围环境的传质. 在此,从催化剂设计原理出发总结了经常使用的提升TMPs析氢性能的调节电子结构和优化次级结构两大策略.
3.1 电子结构的调节
通常,固有的催化活性取决于活性位点的吸附能力和电催化剂的电导率. 合适的吸附能可以有效降低催化过程的过电位,理论上常从电子构型和能带结构解释最佳的吸附能产生的原因. 而更好的电导率则可有效地将电子从电极传输到反应界面,大大加速电催化过程,研究人员又从调整带结构的角度说明电子导电能力的起源. 可见,吸附能与电导率是催化剂电子构型的外在表现. 如若达到提升催化活性点的本征活性,需从电子结构的改变开始. 元素掺杂与界面调节是两种调节催化剂电子结构的常用策略.
3.1.1 元素掺杂 通过其它外来元素掺入TMPs形成掺杂是提高催化剂催化活性的有效策略之一,其它原子的掺杂能破坏原有晶格的周期性,从而改变局部电子结构,达到调节d带中心,优化中间体的吸附能以及提升催化剂的寿命等目的[82]. 杂原子掺杂可大致分为非金属元素掺杂和金属元素掺杂两类.
非金属掺杂一般指O,S,N 等,由于HDS 和HER 有相似的机制,表面的磷硫化物可能对HER 有益. Jaramillo 等[83]便将硫引入磷化钼(MoP)中制备出掺S 磷化钼(MoP|S),MoP|S 催化剂在酸性介质中对HER具有极好的活性和稳定,在电流密度为10 mA/cm2处,过电势仅为86 mV,可解释为S和P相互调节电子特性,产生比纯硫化物或磷化物活性更高的催化剂相,从而促进了HER性能. 且非金属S的存在降低了TMPs 中表面的氧化程度,从而延长了其使用寿命. 在MoP|S 暴露于空气或HER 催化后,未显示P或S. 类似地,Lee 等[84]将S 掺入CoP 中,制备了双功能电解水催化剂S:CoP NPs. 对其HER反应而言,该催化剂在1 mol/L KOH下,在电流密度10 mA/cm2下的过电势为175 mV,优于未掺杂S 的CoP 纳米颗粒,且当电流密度大于50 mA/cm2时其过电势低于商用Pt/C 催化剂. 通过总态密度(TDOS)计算,S的掺入使得CoP的导电能力增强,且使得CoP中Co3d轨道处电子密度更加集中,有利于水的分解[图10(A)~(C)].

Fig.10 iR-corrected polarization curves during HER in 1.0 mol/L KOH for S:CoP NPs(A),total density of states(TDOS)and projected density of states(PDOS)for CoP(011)surface(B,C)[84]
金属掺杂指再在TMPs中引入第2种(或多种)金属元素来调节其局域电子结构. Chen等[85]在Ti网上制备了含Mn 的CoP 纳米片,其在宽泛的pH(pH=0~14)范围内均具有良好的HER 性能. Mn-Co-P/Ti在0.5 mol/L H2SO4,1.0 mol/L KOH和1.0 mol/L PBS中,其η10处的过电势分别为49,76和86 mV,DFT计算表明,Mn 掺杂会削弱Co 和H 原子之间的相互作用,从而导致氢吸附自由能更接近零[图11(A)和(B)].
Liu等[86]从理论上预测掺杂Zn的CoP可能是HER的优良电催化剂,并通过实验验证了Zn0.08Co0.92P纳米壁阵列在酸性和碱性介质中均是性能较好的HER 催化剂. Zn0.08Co0.92P 仅需39 和67 mV 的过电势即可分别在0.5 mol/L H2SO4和1.0 mol/L KOH 达到10 mA/cm2的电流密度. DFT 计算表明,Zn 掺杂使Co-Co桥位有利于氢吸附,由于Zn具有较低的电子负性,因此其可以向相邻的P原子提供一些电子,并产生一些缺电子的阳离子. 为了补偿电子缺陷,周围的Co会损失更多的电子,从而削弱Co原子与吸附的H之间的键合强度. 这一想法可以通过计算d谱带中心的态密度(DOS)得到验证[图11(C)~(F)].
3.1.2 界面调节 合理设计多组分异质结构可显著增强电催化活性. 因为每个组分相之间都具有直接的物理接触,所以界面处的物理化学性质将与本体大不相同. 使得其表现出强界面相互作用,该界面可通过调整界面电子结构来提高电导率,优化吸附能,从而加快反应动力学,最终促进了析氢反应的进行[87].
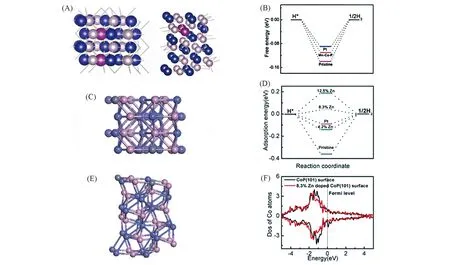
Fig.11 Top and side view of Mn-Co-P(101) with two Mn atoms replacing the subsurface Co atoms(A),free-energy diagram for HER on Mn-Co-P(101)with 8.3%(atom fraction)Mn doping(B)[85], top view(C) and side view(E) of 8.3% Zn-doped CoP, free energy diagram for HER on pristine CoP(101), CoP(101) with 4.2%, 8.3% and 12.5% Zn doping(D), calculated DOS of surface Co atoms on the pristine CoP(101)and CoP(101)surface with 8.3%Zn(F)[86]
其中,TMPs与其它功能材料结合形成异质结构是提高催化剂HER性能的一种策略. 碳基材料是使用最广泛的材料之一,其不仅可以提高混合催化剂的电导率,防止TMPs 氧化,还可以调节ΔGH*.Feng 等[88]在碳纤维支撑的CoP 杂化纳米线(HNWs)上修饰以聚苯胺(PANI)纳米点得到PANI/ CoP HNWs-CFs,在酸性溶液中该催化剂表现出超高的HER 催化性能[图12(A)~(C)],在过电势大于66 mV时其电流密度优于商用Pt/C,在0.02~0.20 V过电势区间中Tafel斜率仅为34.5 mV/dec. 考虑到N原子上大量的孤对电子,PANI会从水合氢离子中捕获H+形成质子化胺基,该质子化胺基的正电荷密度高于水合氢离子,质子化胺基团中的H+能够更好地从阴极接收电子,这对HER 有利. 此外,PANI/CoP HNWs-CFs 的ΔGH*(-0.087 eV)接近零,而CoP 和PANI 的ΔGH*分别为-0.155 和1.168 eV,证明了PANI的修饰优化了氢吸附自由能.
Guo 等[89]通过NiS2纳米八面体磷化构造了空心的Ni2P-NiP2. 从TEM 照片[图12(F)]可见,Ni2P 的(111)面和NiP2的(220)面之间存在明显界面. 当用作HER催化剂时,Ni2P-NiP2表现出优异的电催化性能,电流密度为10 mA/cm2时仅需59.7 mV 的过电势,且Tafel 斜率为58.8 mV/dec. Ni2P-NiP2多晶型催化剂的HER性能均优于Ni2P空心纳米颗粒和NiP2空心纳米颗粒,以及商用固体Ni2P纳米颗粒. 随后,理论计算揭示了界面效应在Ni2P-NiP2异质界面中的重要作用[图12(D)~(F)]. 显然,与Ni2P(-0.326 eV)和NiP2(-0.290 eV)相比,Ni2P-NiP2(-0.161 eV)上有更适度的氢吸附自由能. 且在界面区域发生了强电荷的重新分布,Ni2P-NiP2界面附近的P(NiP2)位的平均价电荷从5.22 eV变为5.05 eV,这可能有助于氢从P位解吸(Ni2P-NiP2),避免催化剂中毒. 这使Ni2P-NiP2多晶型催化剂优化了价电子态且增强了其电导率,从而表现出更好的电催化活性.
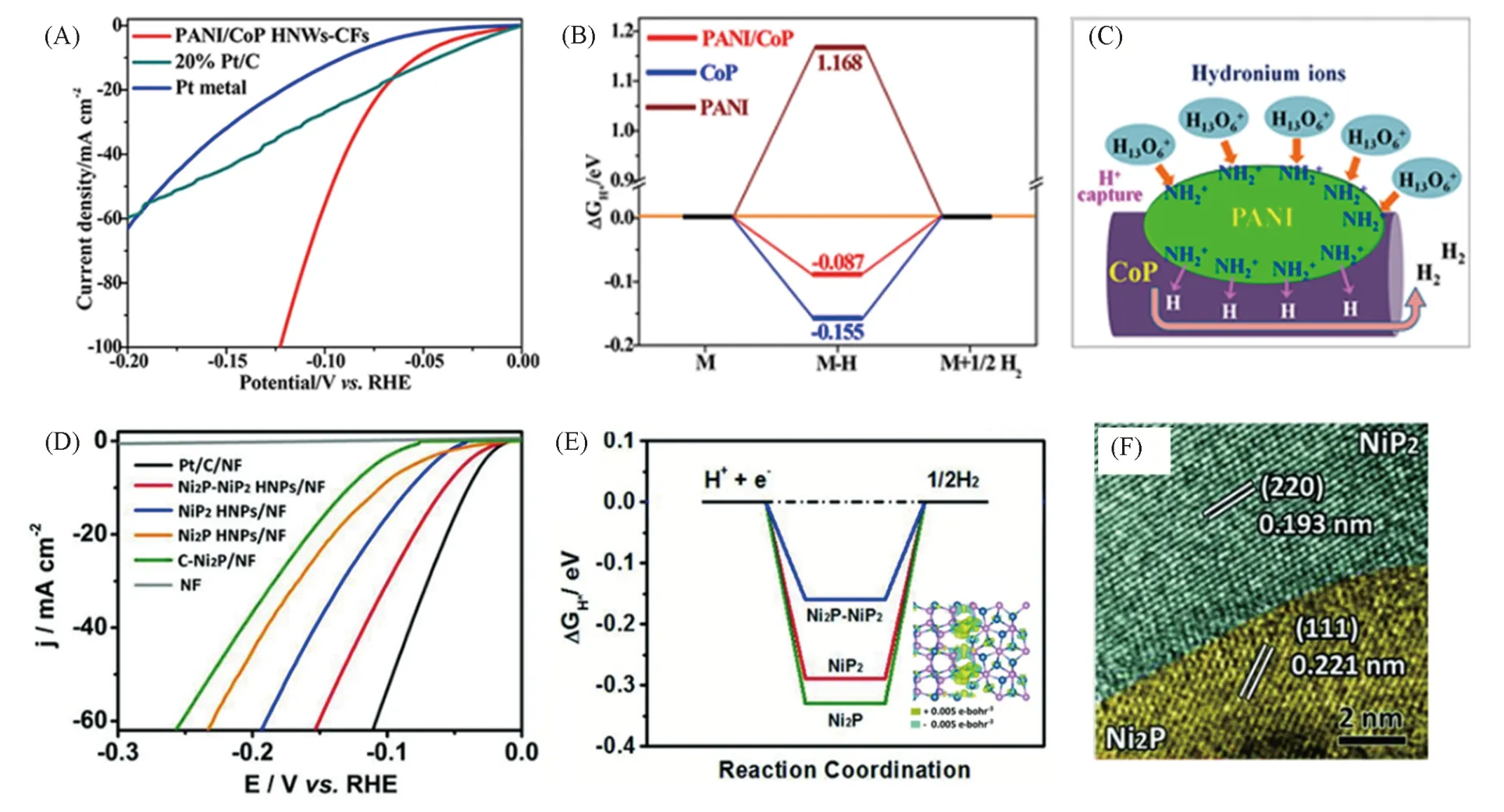
Fig.12 Polarization curves of PANI/CoP HNWs-CFs(A), free energy profile of H adsorption on the different catalysts(B), scheme of HER improvement by capturing the H+ in hydronium ions to form the protonated amine groups on PANI/CoP HNWs(C)[88],HER polarization curves of the Ni2P-NiP2 HNPs/NF(D), HER free-energy diagram calculated at the equilibrium potential for the Ni2P-NiP2 polymorph and charge density difference plot at the Ni2P-NiP2 interface(E),HRTEM image of the Ni2P-NiP2 HNPs(F)[89]
同样地,Ren等[90]通过化学气相沉积法在管式炉中合成了FeP/Ni2P,发现了相对于单一结构,FeP/Ni2P对于HER更具优势,通过DFT计算表明,FeP/Ni2P异质结构使得ΔGH*更为接近零.
3.2 结构工程
在纳米催化中,时刻存在着电荷传输与物质传输,构建合理的催化结构有助于提升催化剂与电解质之间的电荷和物质传递速率,改善其反应动力学. 此外,催化剂的微观结构不同,表明可用的活性位点的数量不同. 从几何因素设计角度出发,也可对TMPs催化剂进行结构上的设计.
通常,经过合理选择能除去前体中的某一组分,使得最终的催化剂具有纳米多孔结构. 如Cheng等[91]通过使用碳球作为自模板,然后进行两步热处理,过程中碳氧化后分解,最终合成了多孔的多壳Ni2P空心微球. 经表征此纳米多孔微球具有大量的2~25 nm之间的空隙. 这种独特的结构使其具有丰富的活性位点和较短的电荷传输距离. 在1 mol/L KOH中10 mA/cm2的电流密度下,Ni2P空心微球仅需要98 mV过电势即可进行HER反应,优于固体Ni2P微球和Ni2P纳米粒子[图13(A)~(C)].
随着电解的进行,气泡将逐渐从催化剂表面产生,而大量吸附在催化剂表面上的气泡将阻碍电催化剂与电解质之间的接触,这将大大减小电化学活性面积[92],从而降低了电催化剂的活性,为了反应的持续高效进行,生成的气泡应该得到及时的脱除[6]. Zhang 等[57]合成了在Ni 泡沫上垂直生长的Ni2P纳米片. Ni2P纳米片的疏松排列和Ni泡沫的网状骨架有利于小气泡的逃逸. 疏水表面降低了产生的气泡的附着力,从而快速去除小气泡保持了活性位点的洁净,从而提高了HER 的效率. 出于相同的考虑,还可通过气泡电镀技术获得多孔的非晶态的过渡金属磷化物,一方面无定型非晶态会形成更多的催化位点,另一方面多孔的形态更有利于之后电解产物的脱附. 如Zhao 等[93]通过气泡电镀技术在镍网NM 上一步制得NiFeCoP/NM,其在1 mol/L KOH 中10 mA/cm2的电流密度下的过电势仅为33 mV[图13(D)~(F)].
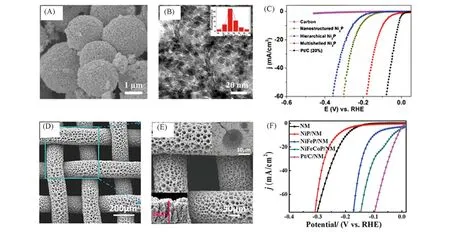
Fig.13 Scanning electron micrograph of the Ni2P particles(A) and high-magnification TEM image of the shell of a sphere(B), linear sweep voltammetry(LSV) polarization curves of multishelled Ni2P(C)[91], SEM images of the NiFeCoP/NM with the low(D) and the high magnification(E),iR-corrected LSV curves for NiFeCoP/NM(F)[93]
此外,还可将TMPs与碳纳米管等碳材料结合,此类方法一方面可使电子传输更为迅速,另一方面其碳材料基底可起到支撑分散活性催化剂的作用.
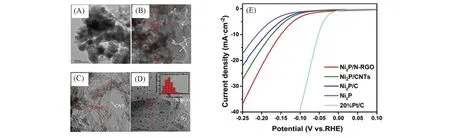
Fig.14 TEM images of Ni2P(A), Ni2P/C(B), Ni2P/CNTs(C) and Ni2P/N-RGO(D), LSV curves of Ni2P,Ni2P/C,Ni2P/CNTs and Ni2P/N-RGO(E)[94]
Qi等[94]分别以C,CNT和N-RGO为基材合成了Ni2P/C,Ni2P/CNT,Ni2P/N-RGO[图14(A)~(E)],并与无基底的Ni2P 对比,发现以上述碳材料为基底的Ni2P的HER性能均优于无基底的Ni2P,从其TEM照片可以看出,结合碳基材后Ni2P 能够得到有效的分散.
以上综述了TMPs 的析氢性能提升的策略(Scheme 2). 从上述多例的优化结果来看,虽然从催化剂设计出发有两大方向,但是,性能提升策略的主要目标应是提升其活性位点的本征催化活性,而不是几何构筑优化传质,这是因为前者的优化对最终催化性能的贡献值常常高于后者,应在提升活性位点的本征活性的基础上继而提升其有效数量,再通过合成合理的结构使得其动力学过程阻力下降,最终使得催化剂性能得到极大改善.
Scheme 2 Schematic diagram of improvement strategies for hydrogen evolution of transition metal phosphates
4 总结与展望
本文总结了过渡金属磷化物在电化学析氢方面的研究进展. TMPs 已被证明是在宽pH 范围内活性、稳定性和成本效益优良的HER电催化剂之一. 首先简介了过渡金属的发展历史以及其基本结构性质和合成方法,然后介绍了过渡金属磷化物在HER中的应用,其中探讨了P含量对过渡金属磷化物性能的影响. 最后介绍了改善过渡金属磷化物HER性能常用的一些方法策略,其中,元素掺杂目的是将外来原子掺入TMPs晶格中以调节电子结构,从而获得最佳的氢吸附自由能. 界面调节旨在实现不同组分之间的协同作用. 两者皆出于调节电子结构的目的,而结构工程主要通过形成多孔结构或选择合适的基质以暴露足够的活性位点,以提升催化剂性能. TMPs 作为优良的HER 催化剂正在快速发展,但是其中也存在着一些问题. 目前,研究大都专注于合成具有新形态的新磷化物并表征其电化学性能,忽略了本质机理和实际应用的探究,但是我们认为两方面的研究均值得关注.
首先从总结、完善过渡金属磷化物体系的角度,需要更加深刻地理解TMPs在HER过程中的反应机理:(1)表面的变化与真正的活性物质. 在催化过程中,所产生的氢的吸附可影响催化剂表面上的原子间力,导致催化剂表面状态的改变. 所以在确定活性位点之前,当务之急是了解催化剂的结构演变,并在电催化过程中识别真正的活性物质. 由于中间物种不稳定,人们对此可能会产生错误的认知. 因此,迫切需要强大的测试表征技术. 如原位拉曼分析和X射线吸收光谱,以揭示这些电化学过程中的真实界面结构和反应活性中间体. 但是,在HER过程中,巨大的法拉第电流和大量的氢气泡使测量变得困难. 很少有研究关注HER 过程中TMPs 表面的变化与真正的活性物质.(2)暴露晶面.TMPs上的催化反应发生在催化剂的表面,而不同的晶面上原子排序有所不同,这意味着不同表面的电子云密度不同,所以催化剂某些晶面的氢吸附自由能对HER并不适合. 因此,为了探索和控制原子在催化剂表面的排列,暴露具有特定催化效应的晶面,对HER具有重要意义. 但是,目前,大多数正在研究的TMPs都是非晶或多晶的. 具有特定暴露面的TMPs的合成仍然是巨大的挑战. 因此,非常需要合成具有特定晶面的TMPs纳米晶体,并进一步研究其对HER的催化效应.(3)理论计算. DFT计算对于设计和预测新型催化剂的催化性能越来越重要. 对于TMPs,目前理论计算可以解释TMPs 晶体结构、化学组成和催化表面电子状态对析氢性能的影响、掺杂其它元素可以改善其活性的原因和TMPs异质材料之间如何发生相互作用. 但仍然存在一些问题,如理论模型均为理想的单晶表面,而实验中TMPs催化剂是多晶的,这使得有时模拟结果与实验性结果有所不同.
从实际应用角度,现阶段过渡金属磷化物距离应用工业电解生产还有相当的距离. 以下几个方面不容忽视:(1)原料和工艺. 过渡金属磷化物要大规模生产,成本控制和环境友好是必须考虑的因素.另一方面,仍需要开发更加低能耗、高产量、可连续的生产工艺.(2)抗电流冲击性. 目前电解水的趋势是利用风能、太阳能、潮汐能等可再生能源为电力来源. 其具有间歇性和不可控性的缺点. 需要催化剂能够承受间断性的电流冲击,不仅对催化剂的使用寿命提出了更高的要求,也需要催化剂能高效、迅速地将水分解(欧盟规定电解水电解器的制氢响应时间在5 s之内).
总而言之,最近的大量研究证明了TMPs在HER催化中优异的性能. 尽管目前还面临着巨大的挑战,但可以预期的是,通过理论计算与大量实验的结合,将来必定会出现高效、廉价的商用TMPs 催化剂.
猜你喜欢
电镀与精饰(2022年10期)2022-10-14
电镀与精饰(2022年3期)2022-03-14
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
电镀与精饰(2021年4期)2021-05-17
中国有色金属学报(2018年2期)2018-03-26
石油化工腐蚀与防护(2017年1期)2017-08-15
中南大学学报(自然科学版)(2016年2期)2017-01-19
中国资源综合利用(2016年7期)2016-02-03
