
二甲双胍通过AMPK/FoxO3a信号通路对创伤性脑损伤后炎症因子的调节作用
2021-05-29 08:16包海军
科学技术与工程 2021年11期
王 钊,包海军
(徐州医科大学基础医学院法医学教研室,徐州 221000)
创伤性脑损伤(traumatic brain injury, TBI)后神经细胞的损伤与修复一直是脑外伤学科领域的热门课题,它不仅包括由直接外力导致的原发性损伤,还包括原发性损伤引起的活性氧应激,神经细胞的萎缩、肥大,水肿,炎症等继发性损伤[1-2]。创伤性脑损伤后自噬标志物-自噬微管关联蛋白轻链3(LC3)增加,自噬受损可能导致神经细胞的损伤[3]。腺苷单磷酸活化蛋白激酶(AMPK) 是对物质和能量代谢敏感的丝氨酸/苏氨酸蛋白激酶[4]。脑外伤后能量合成受到抑制,使用药物5-氨基-1-β-D-呋喃核糖基-1H-咪唑-4-甲酰胺(AICAR)或二甲双胍后促进AMPK激活,对小鼠脑损伤后的空间记忆有显著改善[5]。TBI后的小鼠表现出认知功能障碍和神经胶质细胞的激活,通过敲除受体相互作用蛋白(RIP3)可以促进小鼠海马中的AMPK磷酸化的激活,从而降低了星形胶质细胞的氧化应激、炎症和凋亡[6]。TBI引起的氧化应激,破坏脑能量稳态,并促进神经炎症,这进一步导致了神经元退化和认知功能障碍[7]。因此AMPK的激活可能是治疗TBI后继发性损伤炎症的一个潜在的治疗靶点[8]。
有研究表明二甲双胍可以通过抑制线粒体呼吸链复合物Ⅰ调控AMP/ATP,促进LKB1蛋白激酶的磷酸化进而激活AMPK[9]。TBI导致脑水肿、血脑屏障破坏、神经功能缺失、前庭运动功能障碍和AMPK磷酸化水平降低。应用二甲双胍后可减轻大鼠脑水肿、改善血脑屏障和前庭运动功能障碍。二甲双胍治疗组p-AMPK/AMPK比值升高。复合物C组通过抑制AMPK破坏二甲双胍的神经保护作用研究提示二甲双胍通过磷酸化AMPK抑制TBI介导的继发性损伤,改善脑损伤后神经行为功能[10]。还有研究表明二甲双胍在慢性TBI后至少有5天的治疗时间窗,在此期间,二甲双胍以AMPK依赖性的方式促进组织修复、抑制神经胶质瘢痕形成和小神经胶质的激活[11]。
临床报道表明二甲双胍除了具有降低血糖的作用外,对中枢神经系统相关疾病表现出强大的抗炎活性。结果表明,二甲双胍能显著改善脑损伤大鼠的神经功能缺损、脑水肿和神经元凋亡。此外,二甲双胍可以抑制小胶质细胞的激活,减少促炎细胞因子的产生,包括肿瘤坏死抑制因子(TNF-抑制因子)、白介素1(IL-1)和白介素6(IL-6)抑制因子[12]。最近的证据表明AMPK/FoxO3a轴在细胞周期阻滞、凋亡和迁移中起关键作用[13]。在缺氧诱导的H9C2细胞中,FoxO3a(叉头蛋白O的亚型3a蛋白)表达上调,应用AMPK特异性激活剂AICAR之后研究发现AMPK的激活促进了FoxO3a的表达以及自噬相关基因ATG家族的转录[14]。脂多糖诱导的急性肝衰竭的实验中发现AMPK上调FoxO3a的表达激活自噬,能够改善急性肝损伤,抑制炎症的发生[15]。
综上所述,AMPK的激活有利于应对TBI后脑的能量危机,并具有一定的抑制炎症的作用,FoxO3a的激活能够直接促进自噬相关基因ATG家族的转录和翻译,二甲双胍以直接或间接方式激活AMPK。现利用蛋白印迹实验和荧光免疫学方法探究二甲双胍是否通过AMPK/FoxO3a信号通路途径来发挥对脑外伤炎症的调控,验证二甲双胍这一药物除了可以降低血糖作用外,还发挥对炎症因子的抑制作用,从而开拓二甲双胍药理学作用价值,为二甲双胍的临床用药打下实验理论基础。
1 材料与方法
1.1 材料
1.1.1 实验动物
SPF级健康成年雄性C57BL/6小鼠(25~30 g)购自徐州医科大学实验动物中心,动物许可证号[SCXK(沪)2018-0006]。Tek-cre(T003764)小鼠和带有FOXO3A-loxp/loxp序列的小鼠购自南京生物医学研究所(中国南京),动物许可证号[SCXK(苏)2015-0001]。
1.1.2 实验试剂
实验药物:二甲双胍(metformin,10 g)和复合物C(Dorsomorphin dihydrochloride,Compound C,10 mg)购自MCE公司。主要抗体:兔抗小鼠FoxO3a(75D8)、兔抗小鼠P-FoxO3a(Ser253)、兔抗小鼠P-AMPKα(D4D6D)、兔抗小鼠NLRP3(D4D8T)、兔抗小鼠IL-1β(D4T2D)、鼠抗小鼠LC3β(E5Q2K)均购自美国Cell Signaling Technology公司;兔抗小鼠LC3β(18725-1-AP)、HRP-山羊抗兔(二抗)、HRP-山羊抗鼠(二抗)、Coralite488驴抗兔(SA00013-6)、Coralite594驴抗鼠(SA00013-7)均购自美国Proteintech Group公司;兔抗小鼠IL-1β(ab9722)购自美国Abcam公司。组织细胞裂解液(RIPA)、蛋白酶抑制剂(PMSF)和磷酸酶抑制剂均购自江苏凯基生物技术股份有限公司。
1.2 实验方法
1.2.1 基因敲除小鼠的筛选与鉴定
FoxO3a-loxp/loxp序列的小鼠与Tek-cre小鼠交配产生子一代,子一代小鼠通过自交的方法产生子二代。在子二代小鼠中按照以下引物序列对小鼠后代进行PCR基因鉴定:对于FoxO3a引物,正向,5′-CTGATACCGAAGAGCCTTGC-3′,反向,5′-CTTCCAAACAGAACCTTGTCC-3′;对于Tek-cre引物,正向,5′-ATTTGCCTGCATTACCGGTC-3′,反向,5′-ATCAACGTTTTCTTTTCGG-3′。筛选出FoxO3a-loxp/loxp/Tek+的小鼠后,根据徐州医科大学动物护理和使用委员会批准的方案和标准,将小鼠维持在无特殊病原体的环境下生存。
1.2.2 TBI模型的建立
采用3.5%水合氯醛按照每10 g小鼠体重注射0.1 mL的剂量腹腔注射进行麻醉。采用改良的Feeney’s自由落体模型装置制作小鼠TBI模型。骨钻在颅骨冠状缝后2.5 mm,偏离颅骨中线左侧1.5 mm处钻一直径5 mm孔洞,在开颅的过程中保持硬脑膜的完整。自由落体的装置快速撞击硬脑膜,实验中所用的砝码质量200 g,高度30 cm。撞击柱直径5 mm,撞击硬脑膜面的下降高度小于4 mm,撞击接触时间0.01 ms。缝合头皮[16]。伪手术组仅剪开头皮,骨钻去除颅骨暴露硬脑膜,缝合伤口。取损伤侧的大脑皮质,称量并记录。
1.2.3 动物给药
二甲双胍以150 mg/kg的剂量在8:00和20:00灌胃给药,对照组给予同样剂量的生理盐水[17]。持续给药7 d,第8天在相应时间点建立小鼠TBI模型。复合物C组采取腹腔注射的方法,按照20 mg/kg的剂量在TBI前1 h腹腔注射,对照组给予同样剂量的生理盐水[18]。
1.2.4 Western blot检测
取损伤侧大脑皮质,称重并按照10 mL/mg加入组织蛋白裂解提取液。按照细胞组织裂解液:蛋白酶抑制剂:磷酸酶抑制剂=100:1:1(质量比)混合后加入。匀浆,存放在4 ℃层析柜中裂解30 min,13 000 r/min离心15 min,吸取上清液。(BCA蛋白定量试剂盒测定蛋白浓度。加入蛋白上样缓冲液,95 ℃保温15 min以充分变性。-20 ℃保存待用。配制SDS-PAGE凝胶,电泳分析实验结果。
1.2.5 免疫荧光双染
鼠脑整体取出后放入4%多聚甲醛溶液固定 12 h,经20%、30%蔗糖溶液脱水至沉底。冰冻切片,切片厚度20 μm,切片放于-20 ℃保存,使用时复温30 min。在切片上滴加0.3% TritonX-100溶液,10%驴血清封闭。两个一抗(封闭血清稀释)混合后涂片,4 ℃层析过夜。荧光素标记的二抗混合,然后涂片。室温孵育2 h(从这一步开始避光),磷酸盐缓冲液(PBS)漂洗5 min共3次,滴加4,6-二脒基-2-苯基吲哚二盐酸盐(DAPI)染液。封片,避光观察。
1.2.6 数据处理
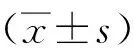
2 实验结果
2.1 二甲双胍对AMPK、自噬及炎症因子表达的影响
二甲双胍组和生理盐水组相关蛋白Western Blot检测结果如图1~图3所示。与生理盐水组相比,二甲双胍组p-AMPK表达增加,p-FoxO3a表达降低[图1(a)、图1(c)]。p-AMPK/AMPK的比值增加,p-FoxO3a/FoxO3a的比值降低,统计学分析数据显示在TBI后的6、12、24 h具有统计学意义[图1(b)、图1(d)]。自噬标志蛋白LC3-Ⅱ/LC3-Ⅰ比值增高,p62表达降低[图2(a)],统计学数据分析数据显示在TBI后6、12 h具有统计学意义[图2(b)、图2(c)]。在TBI后6、12 h,炎症因子NLRP3、IL-1β表达降低[图3(a)],且统计学分析数据显示在TBI后6、12 h具有统计学意义[图3(b)、图3(c)]。由此证明二甲双胍可以促进AMPK的磷酸化,抑制FoxO3a磷酸化,进而促进自噬的发生,对炎症因子起到了一定的抑制作用。

图1 p-AMPK、AMPK、p-FoxO3a、FoxO3a蛋白表达程度及相对蛋白表达量的统计学分析Fig.1 The expression levels of p-AMPK, AMPK, p-FoxO3a and FoxO3a, and the statistical analysis of the relative protein expressions
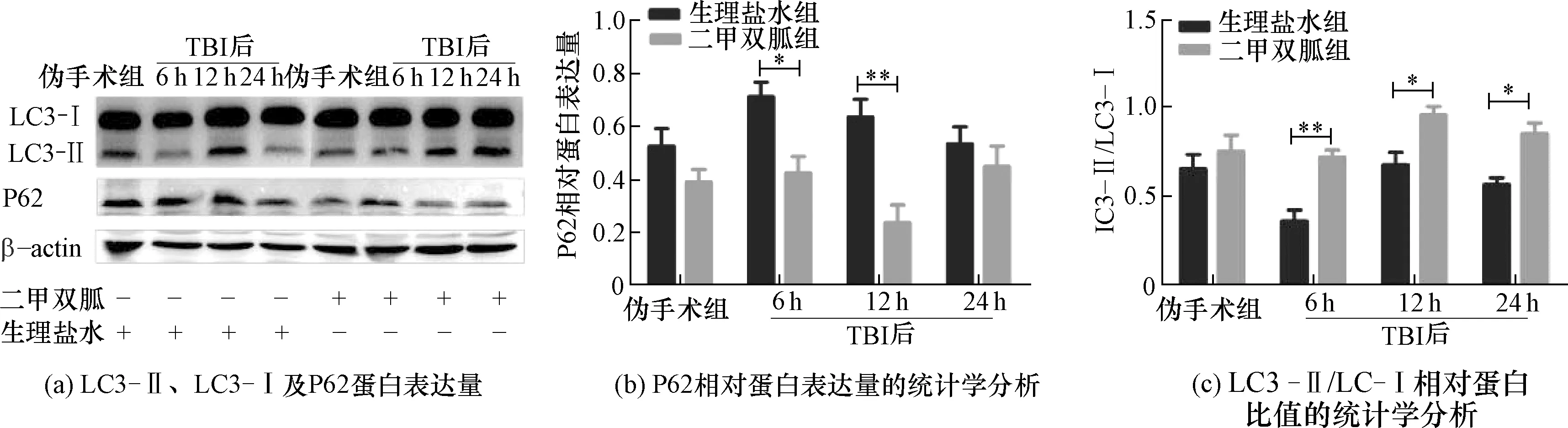
图2 自噬相关蛋白LC3-Ⅱ、LC3-Ⅰ及P62蛋白表达程度及相对蛋白表达量的统计学分析Fig.2 Autophagy related proteins LC3 -Ⅱ, LC -Ⅰ and P62 protein expression level, and the statistical analysis of the relative protein expression

图3 炎症相关因子IL-1β、NLRP3蛋白表达程度及相对蛋白表达量的统计学分析Fig.3 Inflammation of the related factor IL-1β, NLRP3 protein expression level, and the statistical analysis of the relative protein expression
2.2 Compound C对TBI后AMPK、FoxO3a及自噬水平的影响
Compound C 为一种特异性AMPK磷酸化水平抑制剂,选取TBI后12 h的动物模型,建立Compound C组和生理盐水组,实验结果如图4、图5所示。与生理盐水组相比较,使用Compound C后AMPK的磷酸化水平降低,p-FoxO3a的磷酸化水平升高[图4(a)],p-AMPK/AMPK、p-FoxO3a/FoxO3a在TBI后12 h具有统计学意义[图4(b)、图4(c)]。自噬蛋白LC3-Ⅱ/LC3-Ⅰ比值降低,P62蛋白表达水平相反[图5(a)],且数据学分析显示具有统计学意义[图5(b)、图5(c)]。由此证明FoxO3a受AMPK磷酸化水平的调节。p-AMPK表达降低后抑制FoxO3a转录进入细胞核。AMPK活化被抑制后同时抑制自噬发生。
2.3 免疫荧光检测野生型组和基因敲除组TBI后自噬及炎症因子的表达水平
实验结果如图6所示。与野生型小鼠相比较,FoxO3a基因敲除组小鼠自噬水平明显降低,而炎症因子的表达水平明显增高。自噬标志蛋白LC3主要集中分布在细胞质,而炎症因子IL-1β在整个细胞均有分布[图6(a)]。LC3及IL-1β免疫荧光强度采用Image J检测并经过统计学分析显示野生型组和基因敲除组小鼠在TBI后12 h具有统计学意义[图6(b)]。由此进一步证实TBI后FoxO3a能够促进自噬的发生,进而对炎症因子起到抑制作用。
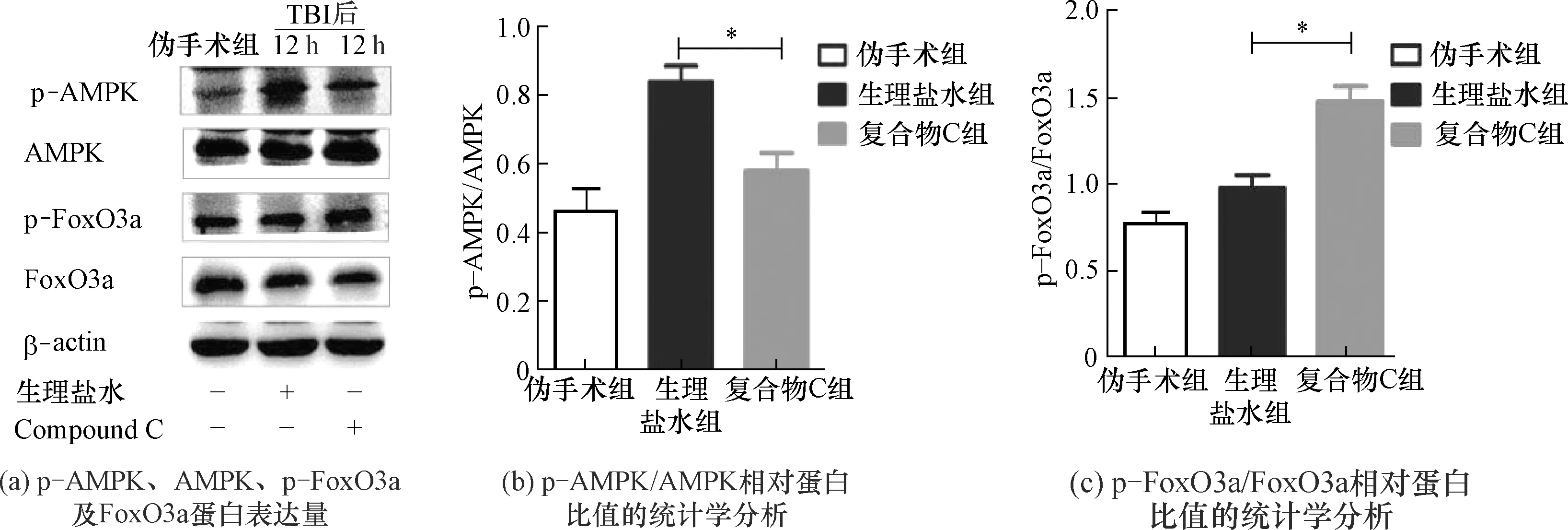
图4 腹腔注射Compound C后p-AMPK、AMPK、p-FoxO3a、FoxO3a蛋白表达程度及相对蛋白表达量的统计学分析Fig.4 The expression levels of p-AMPK, AMPK, p-FoxO3a and FoxO3a after Compound C intraperitoneal injection, and the statistical analysis

图5 腹腔注射Compound C后自噬相关蛋白表达程度及统计学分析Fig.5 Expression level and statistical analysis of autophagy related proteins after Compound C intraperitoneal injection

蓝色代表细胞核;红色代表LC3;绿色代表IL-1β图6 基因敲除组和野生型组免疫荧光染色检测及荧光染色阳性百分比统计学分析分析Fig.6 Immunofluorescence staining detection and statistical analysis of the percentage of positive fluorescence staining in the knockout group and the wild-type group
3 讨论分析
脑内长期代谢抑制是创伤性脑损伤(TBI)的一种典型的继发性病理现象。通过增加能源或提供代替能源的化合物(如三乙酸甘油酯、乙酰左旋肉碱)可以改善预后[19]。AMPK作为一种丝氨酸/苏氨酸激酶,在能量低的状态下被激活[20]。中度至重度创伤性脑损伤会降低AMPK的活性,而损伤后AMPK的激活可以改善空间记忆[21]。AMPK 在 6 个调节位点 (Thr179、Ser399、Ser413、Ser355、Ser588、Ser626) 直接磷酸化 FoxO3a,增强 FoxO3a 转录活性[22]。FoxO3a缺陷小鼠 NF-κB 转录活性明显增强,在 FoxO3a 基因敲除小鼠中,TNF-α诱导的HT29细胞中促炎因子水平显著增加[23]。FoxO3a调节的自噬在发炎的人牙髓和脂多糖处理的 mDPC6T细胞中抗炎作用的研究中发现,FoxO3a细胞核转录的增强与炎症进展呈正相关。FoxO3a可能在维持细胞内稳态和炎症调控中发挥作用[24]。前期相关的课题实验证实了二甲双胍可以通过抑制FoxO3A的磷酸化促 进自噬的发生,进而抑制TBI后小鼠大脑皮质炎症因子的表达[25]。此外在骨骼肌蛋白代谢的调节中发现AMPK促进FoxO3a的转录,进而促进骨骼肌细胞自噬的发生[26]。表明AMPK/FoxO3a途径在多种新陈代谢中起着重要的调节作用。
二甲双胍的抗炎途径除了通过激活AMPK途径,还可以通过抑制胰岛素/胰岛素样生长因子(IGF-1)途径以及NF-κB信号的灭活途径来抑制炎症。在研究老年人急性肺损伤(ALI)的相关实验中发现二甲双胍可能通过AMPK信号通路进而抑制脂多糖诱导下的老龄小鼠ALI中NLRC4炎症小体的激活[27]。同时在脂肪毒性诱导细胞和高脂饮食诱导肥胖大鼠模型研究中发现二甲双胍可通过调节AMPK活性,减少脂肪毒性诱导的细胞间的炎症反应[28]。
通过在体实验的方法探究创伤性脑损伤后二甲双胍对自噬和炎症的调控机制,应用二甲双胍(AMPK的激动剂)和Compound C(AMPK的抑制剂)从正反两方验证AMPK对FoxO3a的调节作用。同时应用FoxO3a基因敲除小鼠免疫荧光双染的方法探究FoxO3a与自噬标志蛋白LC3和炎症因子IL-1β的关系。实验研究结果发现:AMPK的激活和FoxO3a蛋白有利于自噬的发生,自噬的发生起到了对脑外伤后大脑皮质胶质细胞分泌炎症因子的抑制作用。实验结果最终证明二甲双胍通过AMPK/FoxO3a信号通路发挥对TBI后炎症因子的抑制作用,p-AMPK的激活促进FoxO3a的转录翻译,促进自噬体的形成。二甲双胍除了可以降低脑外伤后的血糖异常升高外,对脑外伤后的炎症因子还起到了一定的抑制作用。实验存在的不足之处仅在体实验不能证明二甲双胍直接作用于AMPK/FoxO3a信号通路途径,还需要通过体外细胞培养的实验方法进一步探究。
猜你喜欢
中国临床医学影像杂志(2022年5期)2022-07-26
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
昆明医科大学学报(2021年10期)2021-12-02
科学大众(2021年6期)2021-07-20
昆明医科大学学报(2021年2期)2021-03-29
中华养生保健(2020年4期)2020-11-16
学苑创造·A版(2020年9期)2020-10-13
幼儿画刊(2018年1期)2018-01-04
分析化学(2017年12期)2017-12-25
