
Density functional theory study of formaldehyde adsorption and decomposition on Co-doped defective CeO2(110)surface∗
2021-10-28 07:09YajingZhang张亚婧KekeSong宋可可ShuoCao曹硕XiaodongJian建晓东andPingQian钱萍
Chinese Physics B 2021年10期
Yajing Zhang(张亚婧) Keke Song(宋可可) Shuo Cao(曹硕) Xiaodong Jian(建晓东) and Ping Qian(钱萍)
1Beijing Advanced Innovation Center for Materials Genome Engineering,Department of Physics,University of Science and Technology Beijing,Beijing 100083,China
2School of Mathematics and Physics,Bohai University,Jinzhou 121013,China
3National Supercomputer Center in Tianjin,Tianjin 300457,China
Keywords: first-principles calculations,Co-doped ceria,oxygen vacancy formation energy,formaldehyde dissociation
1. Introduction
Formaldehyde is among the most common and famous indoor pollutants as an important adhesive for wood and wood composites.[1]In the short term,it can contribute to headaches and nausea,irritating the brain,humanity eyes and respiratory tract,but in severe cases,would even damage the liver,the kidney and the nervous systems. Especially in the city,formaldehyde from furniture and mobile homes may cause severe cancer in public.Formaldehyde emission has thus attracted public attention.
Catalytic oxidation is a promising way to eliminate indoor air pollutants. MnO2,CeO2and Co3O4[2]oxides are the more common catalysts. Amongst them, CeO2as one of the conventional catalysts,[3,4]is considered to be a candidate for catalytic oxidation to explore and investigate. CeO2can serve as both catalyst and auxiliary catalytic material.[4–7]Nevertheless, the catalytic efficiency of pure CeO2is desperately low as a catalyst. Its solid solution[8]combined with other metals and doped nonmetals,[9]noble metals[10]and transition metals[11]can enhance the catalytic activity.[12–14]The mixture of Co3O4–CeO2has been extensively examined in the experiment.[15–20]Dhakadet al.[21]found that Co3O4–CeO2mixed oxide catalyst showed an excellent catalytic activity.Doping of CeO2nanoparticles with Co element led to an assistance of the formation of oxygen vacancy and the conversion of Ce3+/Ce4+.[22,23]
Although the experimental studies of doping of Co ions in CeO2has been made, its detailed and validate explanations are still theoretically lacking. What is the essential requirement for the highly catalytic oxidation of CeO2? Compared with pure CeO2(110) surface, high activity of defective Mn doped CeO2(111) surface is due to the high oxidation state of Mn and active surface oxygen species, reported by Hairong Wuet al.[24]But without exotic oxygen species, the full HCHO oxidation reaction can not go on the defective Mn doped CeO2(111). Recently, we investigated HCHO oxidation on the reduced Co-doped CeO2(111)using DFT+Ucalculations.[25]The calculation of transition state of the climbing-image nudged elastic band method (CINEB)based on the Langmuir–Hinshelwood mechanism was carried out, in which both O2and HCHO were co-adsorbed on the CoxCe1−xO2−δ(111)surface. We predicted theoretically that the presence of O2on the surface of the defective Co-doped CeO2(111) led to a reduction in the barrier of the HCHO oxidation reaction. High oxidation activity was attributed to the dopant Co ions and reactive oxygen species, which are in agreement with the finding of the experiment.[26]In recent years,Buet al.[26]reported that the coexistence of Au3+and oxygen vacancy promoted the oxidation of HCHO on Au/CeO2. And Luet al.[27]concluded that plentiful active surface oxygen species played an essential role in the catalytic removal of formaldehyde over Ag/Co3O4–CeO2. Moreover,Nolanet al.[28]and Meiet al.[29]presented that the low-index CeO2(110)was superior to the most stable(111)surface due to more flexible and higher activity.
Based on the above reports, we studied the catalytic activity of removing formaldehyde on defective CeO2(110)surface. We demonstrated the conclusion from Donghai Meiet al.that the oxygen vacancy on the surface of CeO2(110)surface displayed high activity to oxidize HCHO without exotic oxygen species. Based on the Co-doped CeO2(110) surface to be constructed,we calculated the vacancy formation energy of the nearest neighbor oxygen of the Co ions on the surface and the subsurface. We then studied the adsorption energy of HCHO to all the adsorptive sites on the CoxCe1−xO2(110).Finally,the HCHO desorption process was investigated on the defective Co-doped CeO2(110).
2. Computational details and methods
First-principles calculations were performed by Viennaab initiosimulation package(VASP),[30,31]which used pseudopotential in the form of PW91[32]with HubbardU[33]corrected generalized gradient approximation(GGA).[34]The valence electron arrangements of Ce, O, Co, C and H atoms were 5s25p66s25d14f1, 2s22p4, 3d84s1, 2s22p2and 1s1, respectively. The 500eV cutoff energy of the plane wave basis was the same as the previous work.[25]Spin polarization was switched on in all calculations. There was no fixed value ofUto describe precisely all the characteristics of ceria. We setU=4 eV to the description of the Ce (4f) state consistent with the previous work,[25,41]which is included in the range ofU=3–5.5 eV[35]for the reduced CeO2surface. For Co (3d), the value ofU=3[25]was used to better describe the overall electronic structure and surface reactivity.[36]The band gap of bulk CeO2calculated by DFT+U(U=4) was consistent with the experimental value(3.2 eV)and our previous work.[25,37]We chose 7×7×7 Monckhorst–Pack grids for bulk CeO2.The optimized lattice parameters(5.464 °A)of bulk remained the same as the experimental data(5.411 °A).[38]The Hellmann–Feynman forces of each ion were set at 0.01 eV/°A(0.001 eV/°A for bulk)as the standard to reach structural convergence. The threshold of convergence for static calculation of energy was 10−4eV (10−5eV for bulk). We cleaved the relaxed bulk into the p(2×4) supercell CeO2(110) slab of seven layers with the stacking of ABAB···A (see Fig. S1)as our computational surface model,and vacuum thickness of 15 °A was adopted to avoid the interactions. Bottom three layers were fixed to mimic bulk,while the remaining four layers and adsorbed HCHO were relaxed on the CoxCe1−xO2(110)surface. The studies of Wuet al.[24]illustrated that using a larger 3×3×1k-point mesh to optimize the lattice structure was unnecessary. And the samek-point mesh was also used by Meiet al.[29]Consequently, we adopted 1×1×1k-point mesh on theΓ-centered. The climbing image nudged elastic band(CINEB)was used to calculate the energy and transition states of each position of all structures, and its convergence criteria were 0.05 eV/°A.
The oxygen vacancy formation energy was calculated as

whereE[CoxCe1−xO2−δ],E[CoxCe1−xO2]andE[O2]denotes the optimized energy of Co-doped CeO2surface with nonstoichiometry and stoichiometry,and the optimized energy of the free oxygen molecular(O2),respectively.
The adsorption energy of a HCHO molecule on the(110)surface is defined as

whereE[HCHO/CoxCe1−xO2] is the optimized energy of non-stoichiometric Co-doped CeO2surface with the formaldehyde adsorption,andE[CoxCe1−xO2],andE[HCHO],the optimized energy of non-stoichiometric Co-doped CeO2surface and the energy of free HCHO,respectively.
The reaction energy(∆E)and energy barrier(Ea)of each step were calculated according to the following formulas:

whereEIS,ETSandEFSare total energies of the initial state(IS),transition state(TS)and the final state(FS),respectively.
3. Results and discussion
3.1. The oxygen vacancy formation energy on CoxCe1−x O2 (110)surface
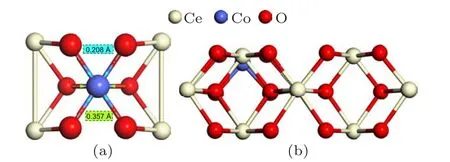
Fig. 1. (a) The close up of Co doping effect in the optimized CoxCe1−xO2 (110) from the top view. (b) Lateral view with only the top three layers of optimized p(2×4)CoxCe1−xO2 (110).
Co doping in the ceria make changes both in the geometric and electronic structure of CeO2(110) surface. As seen from the direction perpendicular to CeO2(110) in Fig. 1(a),the substitution of Co for Ce atom has some visible impacts on p(2×4)(110) surface. Compared with the pure counterpart,NN oxygen atoms centered around Co contracted to dopant by 0.208 °A in the outermost surface of p(2×4)(110),and the similar approach to Co by 0.357 °A for the NNN oxygen atoms in sub-outer layer. Compared with pure CeO2(110), Ce–O length decreases by 0.03 °A in the outermost layer,and the second layer of oxygens in vicinity of Co move up and approximate to the top layer, leading to average reduction of interlayer Ce–O distance by 0.16 °A. In Table 1, we calculate the oxygen vacancy formation energy of NN oxygen and NNN oxygen on the surface and NNsuboxygen on the subsurface for the pure CeO2(110) surface, which are 1.909 eV (NN),1.870 eV (NNN), and 2.907 eV (NNsub), respectively. Also,for the CoxCe1−xO2(110), we obtain the vacancy formation energy of the NN oxygen, NNN oxygen and the NNsuboxygen relative to the Co element, which are−0.725 eV (NN),−0.416 eV (NNN), and 0.259 eV (NNsub), respectively, and the corresponding positions are denoted in Fig. 2. Superior to pure CeO2(110) surface, the doping of Co decreases the oxygen vacancy formation energy, which makes the formation of oxygen vacancies much easier. Likewise, the formation of oxygen vacancy near Co on the(110)surface is easier to form than those on the(111)surface.[25]The negative value of the oxygen vacancy formation energy summarized in Table 1 indicates that the oxygen vacancy at the corresponding positions will be formed theoretically spontaneously. We can find that even vacancy formation energy of the NNsubon the(110)surface requires less energy in comparison to that on the(111)surface. Due to the diversity of low index surfaces, Co dopants have different effects. Obviously,the(110)surface is more active than the(111)surface.Figure S11 showed the projected density of the states (PDOS) of Co-doped CeO2(110)surface. New states have appeared between the valence band(VB) top and the Ce 4f band bottom (compared to the pure surface PDOS). The new states were composed of predominantly down-spin O 2p,Ce 4f and Co 3d states(see Figs.S12 and S13). The reduction of oxygen vacancy formation energy can be ascribed to the gap state decreased, which is consistent with the previous work.[39]The Co doping induced the Ce 4f–O 2p and Co 3d–O 2p gap state acted as electrons acceptor during the oxygen vacancy formation. The introduction of Co into CeO2(110)surface enhanced the catalytic reactivity by adjusting the oxygen vacancy formation energy to a proper value. The oxygen vacancy close to the dopant Co on the surface was easier to form.But NN oxygen on the subsurface(the second layer)was not easy to form vacancy in comparison to NNN oxygen on the surface(the first layer)although NN oxygen was closer to the Co element. Therefore,the existence of V NN on the(110)surface will be our topic for the adsorption of HCHO in the following sections.

Table 1. Oxygen vacancy formation energy for CoxCe1−xO2 and pure CeO2, and eV is the unit of energy. M denotes the spin magnetic moments of Co atom on the CoxCe1−xO2−δ (110) surface, and µB is the unit of M.
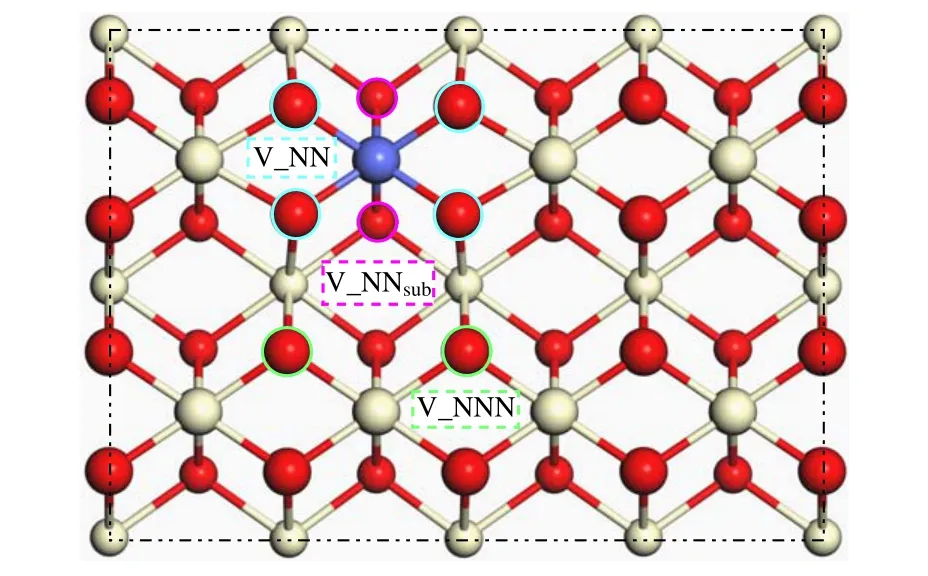
Fig. 2. Two outermost-layer planform of oxygen vacancy on p(2×4)CoxCe1−xO2 (110). We use the cobalt atom (purple ball) in substitution for Cerium atom (white-ivory ball). Take that light cyan circle is the nearest neighbor oxygen(NN)atom relative to Co dopant on the surface(larger ball). V NN means the vacancy of the nearest neighbor oxygen atom when oxygen was removed from surface. The atom in fluorescent green circle is next nearest neighbor oxygen (NNN) relative to Co element. V NNN denotes oxygen vacancy after next nearest oxygen was taken away. The oxygen surrounded by the magenta circle represents the nearest neighbor oxygen of Co on the subsurface (smaller ball). V NNsub is the vacancy of NN atom on subsurface. The same color indicates equal oxygen atom.
The Co ion replacement of the Ce causes its surrounding charge rearrangement on the (110) surface. Nolanet al.[40]reported that the doping of ceria produced two different situations to compensate for the charge,depending on the dopant ion. In the first case,the surface remained stable by the formation of oxygen vacancies.Secondly,oxygen holes and reduced Ce ions (Ce3+) coexisted. Therefore, we computed the electronic density of states of doped ceria(110)surface in order to analyze the Co-doped CeO2(110). The dopant Co resulted in formation of an unoccupied oxygen polaron state,as presented in Fig.S2. The inequality of states for spin up and spin down caused production of magnetic moment.In CoxCe1−xO2(110)surface,Co ion doping resulted in the formation of an oxygen hole to promote the reduction of Ce ion,as shown in Fig.S3.However, we can see that the oxygen atom near dopant Co have no obvious spin polarization.Nolanet al.imposedU=7 for oxygen to describe its visible polarons(noUcorrection to oxygen in this work). We also used HubbardUcorrection in common with the studies of Nolanet al., as illustrated in Fig.S4. It turned out that the NN oxygen of the Co atom has been polarized. This also indicated that the charges of O 2p migrated to Co 3d through the Co 3d–O 2p covalent bonded to generate so-called O 2p holes on the CoxCe1−xO2−δ(110)surface. Our results suggested that doping Co was accompanied by the formation of oxygen hole compensation on the CoxCe1−xO2(110) surface. We do not make further discussion due to beside the point, whether it exists Ce3+or not.Consequently, it can be concluded that dopant Co is accompanied by the spontaneous formation of charges compensating for the oxygen hole on the CoxCe1−xO2(110)surface.
3.2. Adsorption of HCHO over the stoichiometric CoxCe1−xO2 (110)surface
HCHO on the surface of CoxCe1−xO2(110)stabilizes in the tetrahedral adsorption configuration, which is consistent with that on CoxCe1−xO2(111).[25]We can see that the plane configuration of the p(2×4)supercell for CoxCe1−xO2(110)surface was composed of O, Ce (Co) six-atomic rings in the outermost layer and rhombus four-atomic rings in the second outermost layer (refer to Fig. 2). The various adsorption of formaldehyde at different locations of the six-atomic ring were thus summarized systematically in Fig. S5. Most metastable energies of different adsorption configurations varied in the range from−2.76 eV to−2.86 eV in Table S1. The elaborate analysis for all twelve metastable adsorption configurations were given in our supplementary material. From that,we found the configuration with larger adsorption energy has a smaller HCOfbond angle, but a larger HCOsbond angle.Also,we concluded that(e)configuration is the easiest to form and most stable structurally in all metastable configurations(a)–(l) in which C atoms interact with lattice oxygen and O atoms of HCHO bind to Ce atoms on surface.
Compared with formaldehyde adsorption energy on the CoxCe1−xO2(110)surface and(111)surface,it could be seen that the average adsorption energy of HCHO on the(110)surface is higher than the value on the (111) surface, which indicated that (110) surface exhibit better adsorption capacity than(111)surface and demonstrated that(110)surface is more open than (111) surface. In Fig. 3, we presented the most stable configuration and selected strongest adsorption model(Fig. S5(e)) from metastable configurations in Fig. S5, and their structural information of HCHO adsorption are summarized in Table 2,metastable configuration(e),and the strongest adsorption configuration (see Table 2). However, the adsorption strength of configuration with Of–Co bond was stronger than that with Of-Ce. As shown in Fig. 3, we displayed the most stable and metastable configurations and the corresponding charge density differences (CDD). The most stable adsorption configuration with the Of–Co bonding and the C–Osbonding was designated as (Of-Co)-(C-Os) in Fig. 3(a).The distance between the oxygen of formaldehyde and the dopant Co is 1.82 °A, whereas the distance between the C atom of HCHO and the Osis 1.39 °A. The distance of the C–Ofbond in the (Of-Co)-(C-Os) is 1.39 °A, which is equal tod(C-Os). As such, the CH2O2configuration achieves stability on the CoxCe1−xO2(110) surface. The adsorption energy of formaldehyde in the(Of-Co)-(C-Os)is−3.06 eV.The metastable configuration is (Of-Ce)-(C-Os) in Fig. 3(b), the oxygen atom of formaldehyde points to the Ce atom with a distance of 2.91 °A whereas the C atom of HCHO points to Oswith a distance of 1.37 °A.The distance of the C–Ofbond for the (Of-Ce)-(C-Os) is 1.35 °A. The adsorption energy of (Of-Ce)-(C-Os) is−2.93 eV. The CDD illustrated the interaction between formaldehyde molecules and(110)surface. For(Of-Co)-(C-Os),the total charge transfer is 0.89eon the formaldehyde adsorbed on the CoxCe1−xO2(110)surface.For(Of-Ce)-(C-Os), the total charge transfer is 0.86e. These two sites are investigated in our further calculations.
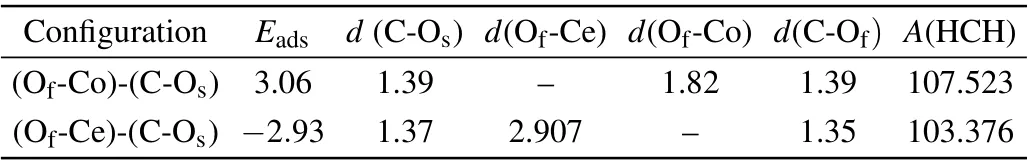
Table 2. Two most stable adsorption configurations, adsorption energies(Eads) (in eV), bond distance (d) (°A) and angles (A) for formaldehyde adsorption on Co-doped CeO2 (110)surface.
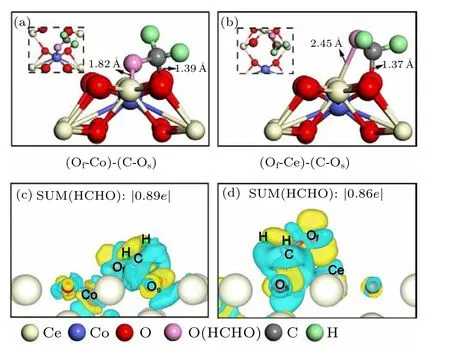
Fig. 3. Representations of most stable (a) (Of-Co)-(C-Os) and metastable(second most stable)(b)(Of-Ce)-(C-Os)adsorption configurations and their corresponding charge density differences(CDD)of(c)and(d)for formaldehyde on Co-doped CeO2(110)surface,respectively. ∆ρdiff isosurfaces were calculated at 0.004e·Bohr−3. The values are effective charges that are calculated by the Bader analysis program. The region of charge accumulation is presented in yellow color,and that of electron depletion in blue color.
4. Formaldehyde oxidation on the defective CoxCe1−xO2−δ (110)surface
Three adsorption modes of HCHO on the CoxCe1−xO2−δ(110) surface were optimized, which were Model-I on the most stable adsorption site, Model-II on the metastable adsorption site, and Model-III on the adjacent oxygen vacancy,respectively In Model-I, the first hydrogen of the adsorbed HCHO dissociated to combine with an adjacent oxygen atom(see IS1~FS1 of Fig. 4). The reaction is 1.1 eV exothermic and has a barrier of 1.05 eV.In addition,the original structure of the second desorbed hydrogen was relaxed. We found that the second dissociated hydrogen ion preferentially adsorbed on the bare oxygen atoms on the surface to achieve stability instead of staying on the adjacent OH∗group. Nevertheless,the barrier energy is up to 3.5 eV (TS1) by calculating the transition states on the bare oxygen atom for the second H dissociative adsorption (see Fig. S6). But the second hydrogen dissociated in the vicinity of the OH∗group required barrier energy of 1.73 eV(see TS2 of Model-I in Fig.4). And also,it was investigated that the barrier energy of dissociative second hydrogen adsorbed on the adjacent OH∗group is only 0.19 eV(see TS2′for Model-I in Fig. S8). The rate-determining step of Model-I is the dissociation of the second hydrogen. The ultimate products of Model-I are H2O and CO2(see Fig.S9(a)).

Fig. 4. Reaction energy profile of formaldehyde oxidation on the CoxCe1−xO2−δ (110)surface corresponding three different original adsorption sites(see Fig.5).
In Model-II,we found that the first dissociated hydrogen has two equivalent adsorption sites, as illustrated in Fig. 5.Firstly, we can see from the top view of the first C–H bond break in model-I-i that the dissociated hydrogen migrates down oxygen. The energy barrier is calculated to be 0.83 eV whereas the reaction energy of this step is−1.56 eV. The second C–H bond cleavage step from the CHOsOf∗group as shown in Fig.5(see IS2~FS2 for Model-II-i)is endothermic(∆E=0.97 eV)and the energy barrier is 1.29 eV.Furthermore,the process of dissociating hydrogen adsorbed on the OH∗group involved an energy barrier of 0.48 eV(see IS2′~FS2′for Model-II-i). As shown in Fig. S9, the final products of Model-II-i are CO2and H2. Furthermore,the cleavage of the first C–H bond to produce the OH∗group and CHOsOf∗group of Model-II-ii requires an energy barrier of 0.91 eV. The C–H bond of TS1 is elongated to 1.13 °A from 1.11 °A. And the step of the second hydrogen adsorbed to produce the H2O∗is the same as the process of the second hydrogen dissociative adsorption for Model-I and Model-II-i. The second hydrogen is dissociated from the CHOsOf∗group,and then dissociative hydrogen is adsorbed on the nearest OH∗group. Their energy barriers are 1.76 eV and 0.60 eV, respectively (see TS2 for Model-II-ii in Fig.4 and TS2′for Model-II-ii in Fig.S8).The final products of the Model-II-ii are H2O and CO2(see Fig.S9(c)).
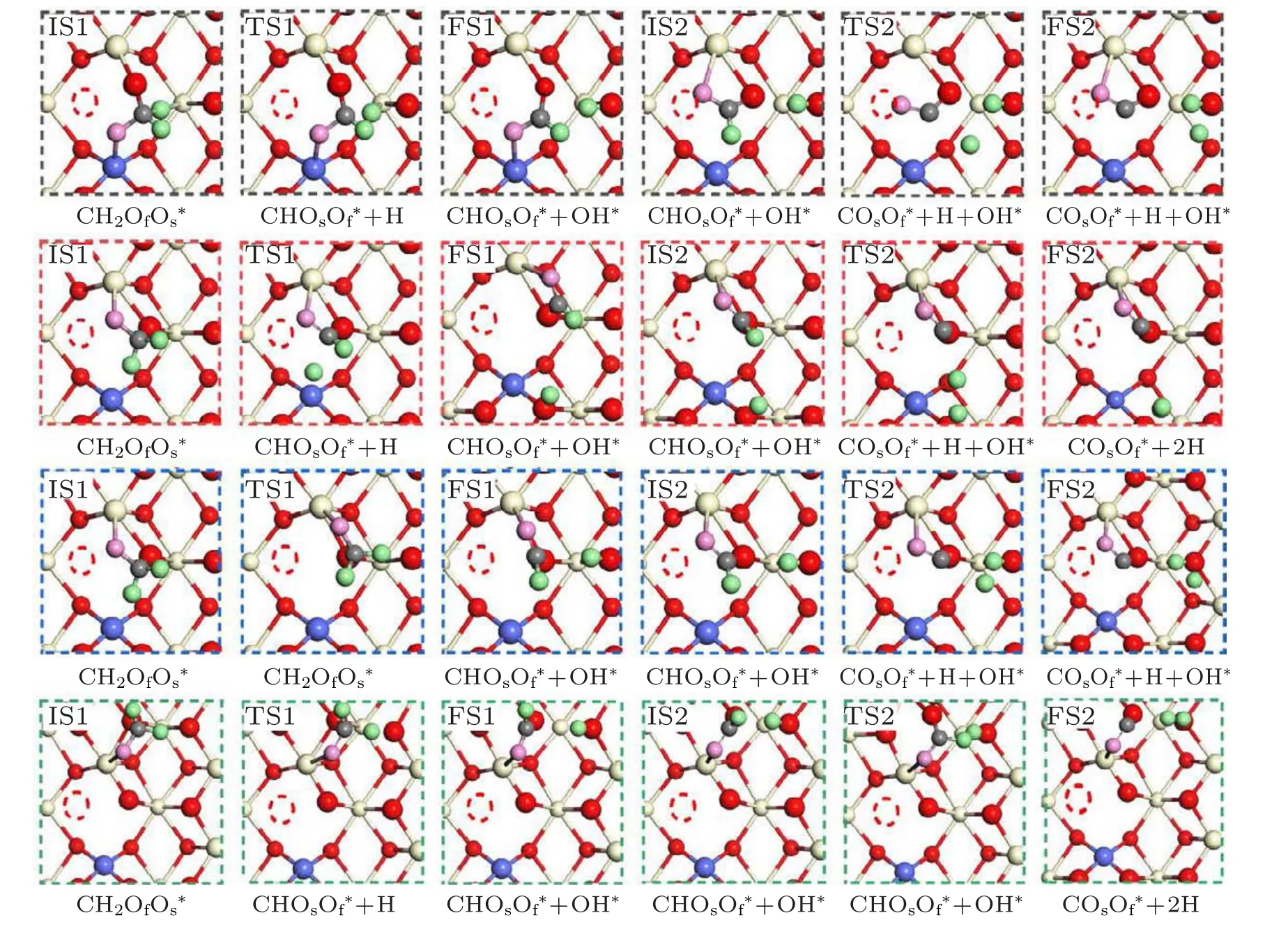
Fig. 5. Four possible pathways for formaldehyde oxidation on the CoxCe1−xO2−δ (110) surface. Of represents the oxygen atom of HCHO molecule, and Os represents the oxygen atom on the(110)surface. The images in black dotted rectangles are Model-I.The images in red dotted line are Model-II-i. The Model-II-ii are in blue circles. The Model-III are in green circles.
From Model-III, the barrier energy of the first hydrogen break at this adsorption site is 0.92 eV.We used the CHOsOf∗group as the initial state for the second C–H bond break in Model-III. Likewise, there are two processes for the second hydrogen dissociation. Deprived hydrogen the CHOsOf∗group gradually became unstable to form COsOf∗and 2H∗.The dissociated second hydrogen was then connected to the OH∗group to produce a H2O∗group and a CO2* group (see Fig. S9). Compared with the initial bond length of 1.11 °A,the C–H bond length of TS2 extended to 1.19 °A. The calculated reaction barriers for the second C–H bond cleavage and dissociated hydrogen adsorption are 1.82 eV and 0.11 eV,respectively. The results revealed that the activation barriers of the rate determining-step for HCHO oxidation on the CoxCe1−xO2−δ(110) surface are related to the distances between the adsorptive sites for oxygen on the surface and dopant Co as shown in Fig. 4 (see IS1 state for three models). The final products of this pathway are CO and H2O(see Fig.S9(d)).
For the three models, the dissociation barrier of the second C–H bond breaking as the rate-determining step is higher than the energy of the first hydrogen dissociation. The Model-II-i exhibits the lowest barrier energy compared to Model-I and Model-II (i, ii), where the rate determining-step is as the judgement. The dissociation energy barrier of the HCHO adsorbed on the oxygen site farther from the Co,i.e.,Model-III,is the highest. Thus,the catalytic activity of formaldehyde oxidation is in the order of Model-II-i>Model-I>Model-II-ii>Model-III.The results indicated that the catalytic activity of the most stable adsorption site is not the highest, while the metastable adsorption site has the best performance for dissociating hydrogen. Therefore, we deduced that the catalytic activity of HCHO oxidation on the CoxCe1−xO2−δ(110)surface does not only depend on the adsorption activity,but also both Co ion and oxygen promote the oxidation of formaldehyde. We also found that the average barrier energy for the second hydrogen dissociation is very high due to the decrease of CoxCe1−xO2−δ(110) surface activity. It might be a better solution to avoid this by introducing exotic reactive species with high activity of oxidation such as O2.[24,25]
5. Conclusion
In summary, the DFT+Utheory was used to study the adsorption and dissociation of formaldehyde on the Co-doped CeO2(110) surface. Firstly, we calculated a series of oxygen vacancy formation energies on different oxygen site centered dopant Co of the CoxCe1−xO2(110) surface. The results showed that the doping Co is accompanied by the spontaneous formation of oxygen vacancies and the oxygen removal of nearest neighbor of Co dopant occurs most easily. Thereafter,we have explored the adsorption energy of HCHO on the CoxCe1−xO2(110)surface. The results revealed that(Of-Co)-(C-Os) model is the strongest adsorption configuration. Finally,we investigated the catalytic activity of formaldehyde on three different initial adsorption states of CoxCe1−xO2(110),and found that the best catalytic performance occurs on the metastable adsorption site, followed by the most stable site.The presence of Co dopant can promote evidently formaldehyde decomposition on defective CeO2(110)surface.

- Chinese Physics B的其它文章
- Physical properties of relativistic electron beam during long-range propagation in space plasma environment∗
- High winding number of topological phase in non-unitary periodic quantum walk∗
- Widely tunable single-photon source with high spectral-purity from telecom wavelength to mid-infrared wavelength based on MgO:PPLN∗
- Control of firing activities in thermosensitive neuron by activating excitatory autapse∗
- Adaptive synchronization of chaotic systems with less measurement and actuation∗
- Dynamics analysis of a 5-dimensional hyperchaotic system with conservative flows under perturbation∗