
Peculiar diffusion behavior of AlCl4 intercalated in graphite from nanosecond-long molecular dynamics simulations∗
2021-10-28 07:02QianpengWang王乾鹏DayeZheng郑大也LixinHe何力新andXinguoRen任新国
Chinese Physics B 2021年10期
关键词:新国
Qianpeng Wang(王乾鹏) Daye Zheng(郑大也) Lixin He(何力新) and Xinguo Ren(任新国)
1CAS Key Laboratory of Quantum Information,University of Science and Technology of China,Hefei 230026,China
2Beijing National Laboratory for Condensed Matter Physics,Institute of Physics,Chinese Academy of Sciences,Beijing 100190,China
Keywords: diffusion,molecular dynamics calculations,graphite,electrodes
1. Introduction
For saving energy and abating pollution,the development of electric vehicles and smart grids becomes an urgent need for the sustainable development of human society, which in turn depends on the availability of low-cost and environmentally friendly energy storage systems.[1]For such applications,the lithium ion batteries(LIBs)are not an ideal option due to the limited availability of lithium in the Earth’s crust. In this regard, the aluminum ion batteries (AIBs) have attached significant attention due to the low cost and safety of aluminum(Al)metal as electrode material. In addition, aluminum ion’s trivalent redox property leads to a higher theoretical capacity of AIBs compared to other rechargeable batteries. The drawback of AIBs for a long time has been their poor cycle and low discharge voltages performance.[2–5]Under such circumstances, the recent breakthrough of Linet al.[6,7]on the Al–graphite cell has drawn much attention. The rate performance of their aluminum-ion battery reached 4000 mA/g when using graphite foam as the cathode, which is a significant improvement over other aluminum-ion batteries.
The excellent performance of the AIB cell of Linet al.[6,7]has attached considerable theoretical interest. Several firstprinciples studies have been performed to elucidate the intercalation behavior of the AlCl4molecule inside the graphite host,[8–18]which is considered to be responsible for the high rate capability and discharge voltages of this AIB cell. In a previous work,[13]we performed density-functional theory(DFT) calculations of the AlCl4graphite intercalation compounds(GICs)to determine the structural and energetic properties of the AlCl4GICs, as well as the voltage of the Al–graphite cell. More importantly, we examined the diffusion behavior of AlCl4GICs viaab-initiomolecular dynamics(AIMD)simulations.From AIMD stuides,we found that there exists a cooperative effect between AlCl4and graphene layers which gives rise to an enhanced mobility of AlCl4. However,our previous AIMD simulation was limited to 20 picoseconds(ps),corresponding to a mean squared displacement(MSD)of~200 °A2for AlCl4and a MSD of~20 °A2for the graphite layers. For a more faithful description of the realistic system,a longer-time AIMD simulation would be certainly desirable.
On the experimental side, graphite has long been known as a good host material for many different guests intercalated in between graphene layers, from small molecules (such as H2[19]) to large organic molecules.[20]Some of intercalation processes (such as AlCl−4, BF−4, PF−6, etc.[10,21,22]) are suitable for half reaction in battery electrodes,[23–25]but the ultrafast charging rate (>1000 mA/g) is only reported on AlCl4GICs when using specific types of ionic liquid electrolyte and graphite cathodes. Considerable efforts have been devoted to examine the performance of various forms of graphite materials as cathode and to elucidate the relationship between the atomic structure and the electrochemical properties. For the graphite cathodes,the early method to obtain ultra-fast charging efficiency was grapheneization.[6,26,27]Later, Wanget al.[28]observed that sonication renders kish graphite capable of supporting a high charging rate (10000 mA/g) while maintaining a high cathode capacity (>120 mAh/g). In addition, Mckerracheret al.found that carbon paper consisting of micron-sized graphite particles still has a capacity over 20 mAh/g even being charging at 20000 mA/g, while pyrolytic graphite foil loses all capacity when being charged over 5000 mA/g.[29]They suggested that GICs in smaller particle size more likely undergo less effect of strain, and thus support a higher charging rate. But previous experimental studies did not clearly identify the characteristic of graphite that can support high charging rates. A computational study of the diffusion behavior of AlCl4in graphite may help us gain a deep insight into this question.
In this work, we set up to run a long-timeab-initiomolecular dynamics (AIMD) simulation in order to examine more thoroughly the diffusion behavior the AlCl4within the graphite host. From this endeavor we find that the diffusion of AlCl4inside graphite and the interaction between the intercalated AlCl4and its host are much more intricate than expected. We find that the diffusion of AlCl4is impeded when the interaction between AlCl4and the graphite host gets weakened,which could occur during a long-time dynamic evolution of the GIC system. It leads to the stagnation of AlCl4and a breaking of the Einstein relation between the mean square displacement (MSD) and the duration time. In addition, we investigate the influence of strain on the diffusion behavior of AlCl4GICs,and find that their presence slows the diffusion of AlCl4and hence is detrimental to the rate performance of the GIC-based AIB.
2. Computational details
As mentioned above, in this work we continue to study the diffusion behavior of an intercalated molecule inside graphite by AIMD simulations. To this end,the atomic-orbital basedab-initiocomputations at USTC (ABACUS)[30–32]package is used. In ABACUS, the norm-conserving pseudopotential[33]in its fully separable form[34]is employed to describe the ionic cores. Furthermore,ABACUS allows to use both plane waves and numerical atomic orbitals(NAO)as the basis functions. In this work, the double-ζplus polarization (DZP) NAO basis set is used, whose adequacy has been benchmarked in previous publications.[13,31,35]In this work,the PBE-D2 functional[36]is used in density functional calculations.
Specifically, we focus on the AlCl4GIC system in its state-4 configuration (i.e., one interlayer gallery being intercalated after every three empty ones). The optimized structure of AlCl4GIC (with one AlCl4molecule in a 4×4×2 graphite cell)is taken as the initial structure in the AIMD simulation. The canonical ensemble (NVT) with Nos´e–Hoover thermostat[37,38]is used and the simulation temperature is set to 300 K. In the present work, the AIMD simulation is performed up to 160 ps with a time step of 1 fs. From the MD trajectory,the diffusion coefficient is calculated by[39]
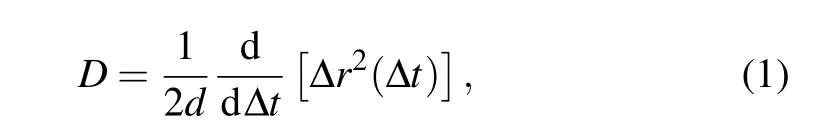
where ∆r2(∆t)is the mean square displacement(MSD)of the moving species as a function of the duration ∆t, anddis the dimension of the diffusion path of the system (d=2 in the present case). The MSD is evaluated from the MD trajectory after the thermodynamic equilibrium is reached and the diffusion coefficient is obtained from the slope of the MSD–∆tcurve as given in Eq.(1).
To accelerate the MD simulations and thus further the accessible timescale, we additionally perform semiempirical quantum-mechanical molecular dynamics using extended tight-binding program package xTB (extended Tight-Binding).[40]This package uses an extended tight-binding method, which allows to treat larger molecular size and/or to run long-time MD simulations, with special attention to the computation of geometries,frequencies,and non-covalent interactions(GFN).[41]The MD parameters(time step,temperature,fictitious mass)used in the xTB-based MD simulations are the same as those in the ABACUS-based AIMD simulation. Since we are using the NVT ensemble, the lattice vectors of the GIC are kept fixed during the MD simulations. A fully relaxed,initially strain-free unit cell of graphite will very likely experience finite strains upon the intercalation of guest molecules.In the xTB MD simulations,two sets of lattice vectors are used.In the first case,the same lattice vectors are used as the ABACUS-based AIMD simulations, and the MD simulation runs up to 1920 ps. In the second case, we adjust the lattice vectors to reduce the strain of the GIC unit cells with respect to the pristine graphite, and run the MD simulations up to 960 ps. In this way,we are able to learn the influence of strain effects on the diffusion behavior of AlCl4GICs.
3. Results and discussion
We first present and analyze the results from the AIMD simulations. In the present work,based on the MD trajectory during a time periodttot, the MSD of a mobile atom or ion is calculated as follows:
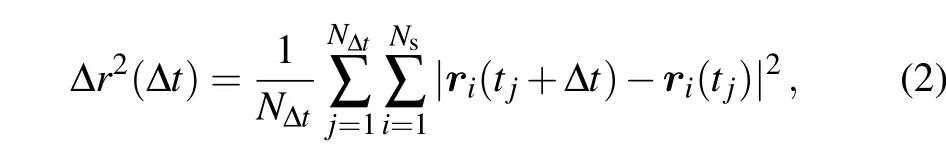
wheretj=(j −1)tstep(tstep=1 fs being the MD time step)is the initial timing of the time interval(tj,tj+∆t). The number of different time intervals with the same duration ∆tis given byN∆t=(ttot−∆t)/tstep,which may get too few for ∆t →ttot. In order to have sufficient displacement sampling,it is suggested in Ref.[42]that the upper limit of ∆tshould not exceed 0.7ttot.Here for safety,we set the maximal ∆t=ttot/2. Furthermore,in Eq. (2),Nsis the number of atoms belonging to the same species in the system. Here, we focus on the motion of the Al atom, which is the center of mass of the AlCl4molecule and hence can well represent the overall diffusion behavior of AlCl4. Since there is only one Al atom in the system, hereNs=1.
In Fig. 1, the MSD data for the Al atom of the AlCl4molecule versus the duration time are plotted. The different MSD–∆tcurves correspond to different choices ofttot(hence different ∆tranges) from a totally 160-ps long AIMD simulations. Specifically,the atomic displacement datar(t)of the first 40 ps, 80 ps, 120 ps, and finally the entire 160 ps of the MD trajectory are employed, and these, via Eq. (2), lead to MSD–∆tcurves with upper limits of ∆tof 20 ps,40 ps,60 ps,and 80 ps,respectively.

Fig. 1. The MSD (in °A2) versus the duration ∆t (in ps) for the Al atom in AlCl4 GICs for different simulation time lengths. The GIC contains one AlCl4 molecule in a 4×4×2 graphite unit cell. The AIMD trajectory is obtained from ABACUS calculations.
From Fig.1,it can be seen that for shorterttot,the MSD–∆tcurves show a good linear behavior,characteristic of typical
diffusion processes. However,asttotgoes beyond 100 ps,the MSD value first drops down, and then rises again; the entire MSD–∆tcurve deviates significantly from the linear behavior.The diffusion coefficients extracted from a linear regression of the MSD–∆tdata,together with theR2parameters of the linear fitting,are presented Table 1. One can see that forttot<80 ps,theR2value is very close to 1,indicating that the linear fitting works well. In consistent with the behavior of the MSD–∆tcurves,when the simulation time exceeds 100 ps,theR2value goes substantially below 1,indicating that the linear fitting to the Einstein relation(Eq.(1))does not work any more. Moreover,from Table 1,one may see that the fittedDvalues have a significant level of statistical variance,even in the time range where the linear fitting still works.This kind of statistical variance stems from the stochastic nature of the diffusion process and has been analyzed in detail in Ref.[42]. However,the behavior of AIMD simulations we have observed,i.e.,the MSD–∆tcurve undergoing from a linear to non-linear behavior,has not been reported and carefully analyzed up to our knowledge.

Table 1. Diffusion coefficients D’s(in cm2/s)of the Al atom in AlCl4 GICs for different simulation times(in ps),extracted from the MSD–∆t data produced by ABACUS AIMD simulations. The goodness of the linear fit(the R2 values)of the MSD–∆t curves are also shown in this table for each time period. The initial 5 ps AIMD steps are excluded from the MSD evaluation in Eq.(2),following Ref.[42].
To understand why MSD values substantially decrease for simulation time longer than 100 ps, we look more closely at how the intercalated AlCl4molecule moves within the host graphite. In Fig. 2(a), we show how thex,y,zcoordinates of the Al atom vary during the whole simulation process up to 160 ps. As excepted,thezcoordinate of the Al atom does not show a noticeable change, indicating the 2-dimensional (2D)nature of the diffusion process. Thex,ycoordinates, on the other hand,change rapidly first,until a time period of roughly 50–70 ps,where a plateau behavior appears. This implies that,during this period, somehow the AlCl4molecule essentially becomes trapped, such that their coordinates show very little variation.After 70 ps,the molecule manages to“escape”from the trap and begin to move rapidly again, until it gets stuck again between 110 ps and 135 ps. Due to the presence of such long(~20 ps)stagnation time,it happens that the MSD value does not increase monotonically as a function of the duration time ∆t. Indeed,we observe that,when the upper limit of the duration ∆tcontains at least one relatively long stagnation period,the decline of the MSD–∆tcurve will appear.
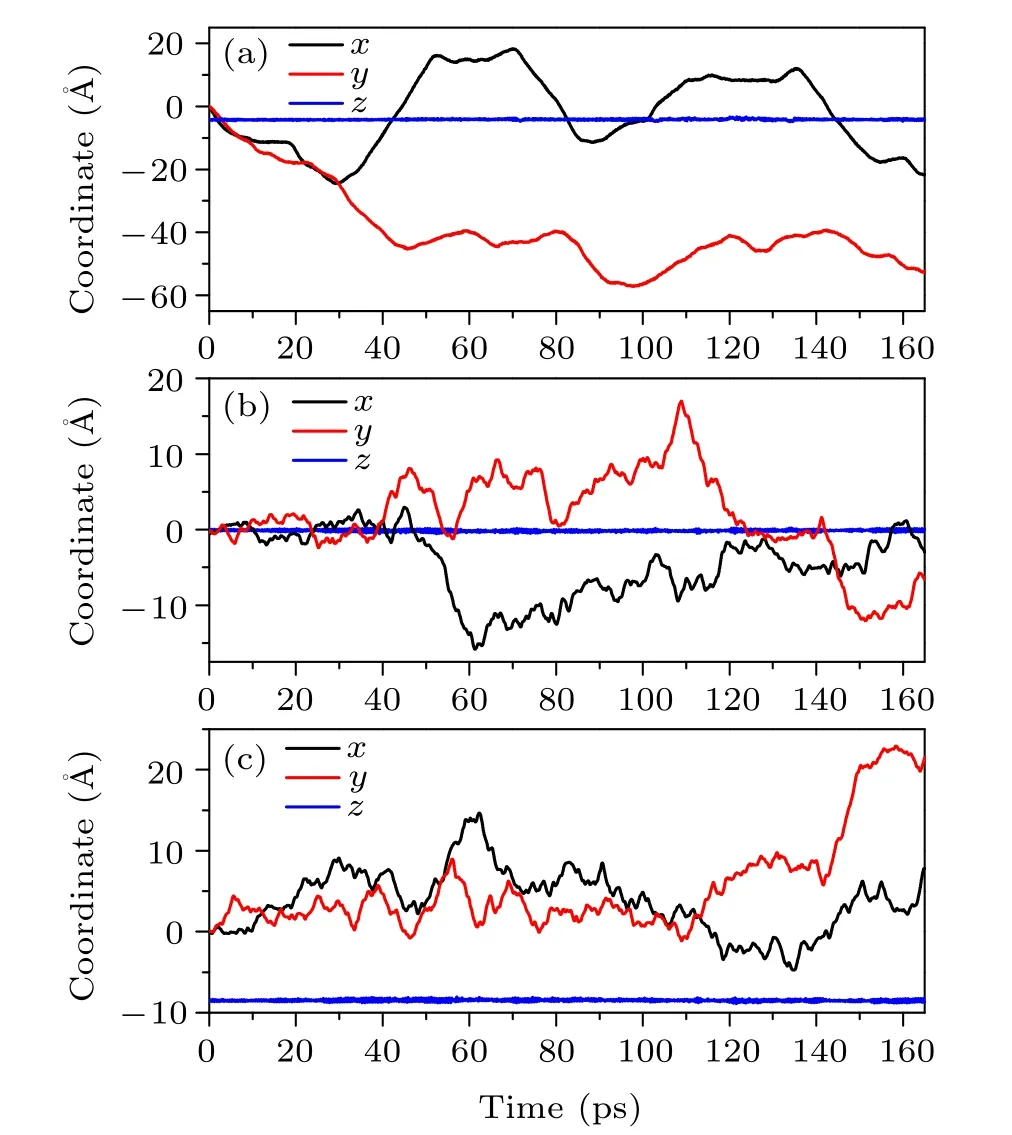
Fig.2. The variation of the x,y,z coordinates(in °A)for Al atom(a)and the center of mass of the upper(b)and lower(c)graphene layers encapsulating AlCl4 as a function of the simulation time(in ps). Data extracted from the ABACUS AIMD simulation.
In order to gain a deeper insight into this peculiar phenomenon, it is necessary to investigate what happens for the entire GIC system when the intercalated molecule gets stagnant. In our previous work,[13]we found that there is a cooperative effect between the intercalated AlCl4molecule and the graphite host, which leads to an enhancement of the mobility of AlCl4. In this work, we again observe a clear correlation between the molecule and the host system. In Fig. 2,thex,y,zcoordinates of the centers of mass of the upper and lower graphene layers encapsulating AlCl4are also presented as a function of the simulation time. One can clearly see that the motion of the graphene layers is accelerated when AlCl4gets stuck around 60 ps and 120 ps,suggesting that there is a transfer of the kinetic energy from the intercalated molecule to its host on these occasions. To have a rough estimate of the strength of the correlation between AlCl4and the host graphite, we compute the binding energies of AlCl4GIC on selected time points along the MD trajectory. Here the binding energy between the intercalated AlCl4molecule and the graphite host is calculated as
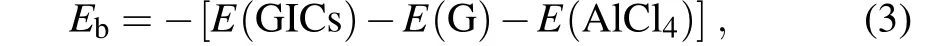
whereE(GICs),E(G), andE(AlCl4) are respectively the total energy of the GIC, the energy of the graphite supercell,and the energy of the AlCl4molecule in their instantaneous structures.Thus a larger positiveEbimplies a stronger interaction between the intercalated molecule and the host.As shown in Fig.3,the instantaneous binding energy fluctuates strongly along with the time. However,if one performs a weighted average over adjacent time steps, the resultant binding energy curve gets smoothed and one can see(on average)a clear decreasing of the binding energy in the time periods around 70 ps and 120 ps. We expect that the decreasing of the binding energy leads to a weakening of the correlation between the movement of the AlCl4molecule and that of graphite,which further weakens the cooperative effect between the two subsystems.When this happens, the diffusion of the AlCl4molecule becomes very ineffective, and the entire diffusion process becomes rather inhomogeneous.
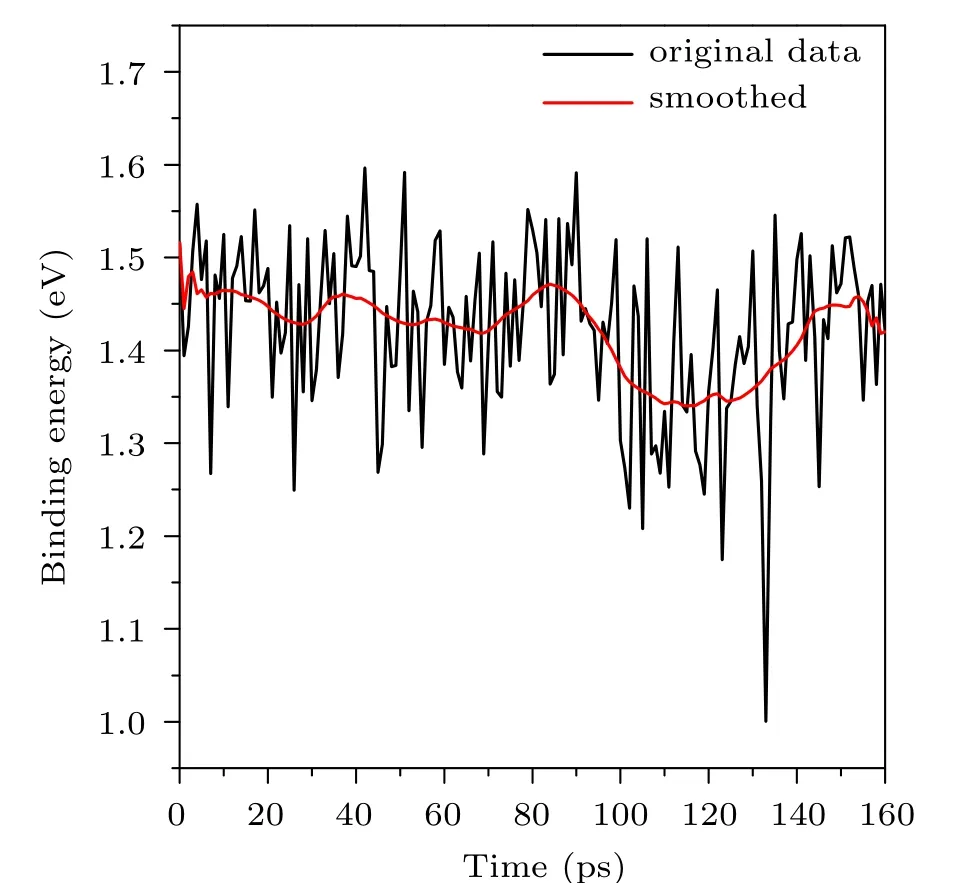
Fig. 3. Variation of the binding energies (in eV) of AlCl4 GICs during the ABACUS AIMD simulation. The original data are obtained according to Eq. (3) every picosecond, yielding 161 data points in total. The red(smoothed)curve is obtained by smoothing over the original data using the weighted adjacent averaging algorithm with the points of window setting to be 20.
The inhomogeneity of the diffusion behavior of the intercalated AlCl4molecule can also be understood via the variation of the diffusion barrier height. To this end,we perform a detailed barrier height study at a few time points along the AIMD trajectory in both regimes (time periods) where the molecule diffuses fast and regimes where the molecule gets stuck. In each regime, we select pairs of two close-by time points(snapshots),and cool down the temperature of the GIC system to 0.1 K in order to get an initial and a final structure.Next,we employ the nudged elastic band(NEB)method[43–46]to calculate the diffusion barrier heights of AlCl4between the initial and final structures obtained above. Specifically, we calculate the barrier heights between five pairs of the(cooled)structures at{24, 25.5}ps,{47, 48}ps,{66, 69}ps,{73,75}ps, and{137, 138}ps respectively. We find that the diffusion barrier height at the time where the molecule gets stuck(66–69 ps) is about 30 meV, whereas its values is about only 7–15 meV at the time where the molecule moves fast. The diffusion barriers are similar to previous calculated values reported in the literature (12–29 meV,[10]21–28 meV,[11]and 8–13 meV[12]).
The phenomenon that the AlCl4molecule gets stagnant occasionally and returns to its usual diffusion behavior after a while can be seen as a “rare event” occurring during the molecule’s diffusion process across the graphite host. For a short simulation time,say below 20 ps,such kind of rare event hardly happens,and the molecule’s movement follows a characteristic diffusion behavior. However,as the simulation time keeps increasing, rare events(the stagnation of the molecule)start to appear, the motion behavior of the molecule experiences a sudden change.In theory,if the simulation time is long enough and samples plenty of periods during which the diffusion of AlCl4is impeded and recovers again, i.e., when“rare events”are adequately accounted for statistically,the MSD–∆tcurve should become a straight line again. By then the diffusion coefficient can be reliably extracted,but it will be different from that extracted from a short-time simulation,due to the presence of the rare event. We conjecture that the peculiar behavior we see in Fig.1 is a manifestation that the simulation is undergoing a transition between two different regimes.To verify this conjecture,it would be necessary to run an extremely long MD simulation. However, this would goes beyond the realm of current AIMD simulations. To proceed,we resort to a much cheaper, but still reasonably accurate semi-empirical quantum-mechanical approach – the xTB model[40,41]mentioned above. Leveraged by the xTB model,we are able to run the MD simulation up to 2 ns.

The MSD–∆tcurves for different total times of the xTBbased MD simulations are presented in Fig. 5. However, a strong non-linearity of the curves is still present, and the extracted diffusion coefficients, as presented in Table 2, vary a lot as the simulation time continues to increase. Generally,the diffusion speed is reduced by one order of magnitude when the simulation time increases from~100 ps to~1 ns. Close inspection reveals that the diffusion behavior of AlCl4in GIC as given by the xTB-based MD simulations is similar to what happens in the AIMD case. Namely, the migration of AlCl4gets stagnant occasionally during the simulation,for instance,during the time periods 380–430 ps,600–650 ps,750–820 ps,920–1020 ps, and 1640–1720 ps. Thus, during the much longer xTB-based MD simulation,the“rare events”indeed occur multiple times,and substantially influence the overall diffusion property of the GIC system.However,it seems that they are still not adequately accounted for in statistically meaningful way,so that the expected characteristic linear MSD–∆tbehavior is not yet achieved. We anticipate that one likely needs to go up to~10 ns total simulation time to see the restored linearity of the MSD–∆t.

Fig.4. The L2 norm of forces(in eV/°A)for AlCl4 GICs as a function of the simulation time (in ps) during the ABACUS (a) and xTB (b) MD simulations.

Fig. 5. The MSD (in °A2) versus the duration ∆t (in ps) for the Al atom in the stage-4 AlCl4 GICs for different simulation time using xTB, different colored lines represent different lengths of simulation time.

Table 2. The diffusion coefficients(in cm2/s)of Al atom in AlCl4 GICs for different simulation time(in ps)obtained from xTB MD simulation.
In our analysis above, we suggested that the impeded diffusion of AlCl4in graphite is connected to their weakened interaction. Experimental study[29]indicated that the strain present in the system may affect the rate property of the graphite cathode of AIB,providing another perspective for understanding the peculiar behavior discussed above. Inspired by this observation,we perform a series of computational simulations to examine the influence of strain on the diffusion behavior of AlCl4in GIC. To this end, ns-long xTB-based MD simulations with different initial structures subject to different levels of strains are performed.Here,the level of strain is measured by the distortion of the optimized lattice vectors upon the AlCl4intercalation with respect to the perfect graphite structure. In the simulations discussed above,the optimized lateral lattice vectors of GIC deviate noticeably from the hexagonal symmetry,in addition to a drastic expansion of the inter-layer distance in thezdirection. To check the effect of strain, we first set up a“strain removal structure”by artificially restoring the hexagonal symmetry of the in-plane lattice vectors, and use this structure as the initial structure for xTB MD simulations. As it turned out, by doing so, the motion of AlCl4is less frequently stagnated,and the diffusion process is more continuous. The resultant MSD–∆tcurve is then plotted in Fig. 6 (red curve), whereby one can see a sizeable enhancement of the MSD,compared to that obtained with the original structure (black curve). This shows that the molecule indeed diffuses faster when the strain is removed.
To further investigate the effect of strain along thezdirection,we set up another simulations with a slab model,with AlCl4sandwiched in between two graphene layers. This slab structure is created by adding a large vacuum to the aforementioned strain removal structure. Compared to the bulk GIC model, the slab structure allows the system to expand freely in thezdirection,ending up with a larger strain in thezdirection and on average a weakened interaction between AlCl4and the host. As expected, the xTB MD simulation based on this structure shows that the AlCl4gets stuck more often during its diffusion,compared to its bulk counterpart,and hence yields a suppressed MSD,as shown in Fig.6(blue curve).On the other hand, if one doubles the magnitude of the strain of the bulk structure inx–yplane,the xTB MD simulation yields a MSD(purple curve in Fig.6)that is much suppressed compared to that of the original structure. All these numerical experiments clearly demonstrate the close relationship between the strain in the system and the diffusion properties of the intercalated molecule. It appears that the strain present in GIC-based cathode is harmful to achieve high-rate properties in AIB.

Fig.6. The MSD(in °A2)versus duration ∆t (in ps)for the Al atom in AlCl4 GICs in 960 ps MD simulations using xTB with different initial structures(lattice vectors). The black, red, blue, and purple curves correspond to the original structure(i.e.,fully relaxed with stress below a threshold),a strain removal structure whereby the in-plane lattice vectors respect the hexagonal symmetry, a slab model with no strain in the x–y plane, and a strain doubled structure where the lateral strain is doubled compared to the original structure.
4. Summary
The diffusion behavior of a molecule inside graphite materials is studied via MD simulations, using the AlCl4intercalated GIC as a prototypical system. Long-time MD simulations reveal that the AlCl4molecule may get stuck during its diffusion in between the graphene layers from time to time,and recovers its normal (fast) diffusion behavior after struggling for a while. As a result, from short-time (a few tens of picoseconds) to long-time (hundreds of picoseconds to a nanosecond) MD, the MSD–∆tcurve changes significantly,rendering a reliable extraction of the diffusion coefficients impractical. Such phenomena are observed in both AIMD and MD simulations based on semiempirical extended tightbinding (xTB) models, and thus reflect the generic feature of such types of systems. We propose that this peculiar behavior arises because the simulations are undergoing a transition regime where the rare event (here the stagnation of the intercalated molecule) starts to appear but is not yet sufficiently accounted for. In such a transition regime, the usual Einstein relation characterizing the typical diffusion process is not fulfilled. Based on our current simulations, we expect that MD simulations of time length of 10 ns or longer are required to pass this transition regime and reach a quantitative assessment of the diffusion properties of AlCl4intercalated GIC.
Close inspection reveals that the stagnation of the molecule is related to the decrease of the average interaction strength between the intercalated molecule and the graphite host, that can appear occasionally due to an intricate interplay between the two subsystems at finite temperature. This will result in a weakening of the cooperation effect of the GIC system, as discussed in Ref. [13]. Furthermore, we also investigated the influence of the strain present in the GIC system on the diffusion properties. Several numerical experiments clearly show that when stronger strains are present,the molecule gets stagnant more often,leading to an overall slower diffusion. We conclude that the strain is detrimental to the rate property of the GIC cathode. More studies along this line are necessary to fully elucidate the problem. Our preliminary results suggest that finding means to reduce the inhomogeneous strain in GIC systems during charging,e.g.,via material engineering,should be helpful to improve the rate performance of the graphite electrode.
猜你喜欢
安顺学院学报(2022年1期)2022-03-17
品牌研究(2022年7期)2022-02-06
宝藏(2021年1期)2021-12-06
新世纪智能(高一语文)(2021年4期)2021-07-28
新世纪智能(高一语文)(2021年4期)2021-07-28
宝藏(2021年3期)2021-04-20
宝藏(2021年1期)2021-03-10
宝藏(2021年1期)2021-03-10
农药科学与管理(2019年5期)2019-08-13
当代经济(2015年4期)2015-04-16

- Chinese Physics B的其它文章
- Physical properties of relativistic electron beam during long-range propagation in space plasma environment∗
- High winding number of topological phase in non-unitary periodic quantum walk∗
- Widely tunable single-photon source with high spectral-purity from telecom wavelength to mid-infrared wavelength based on MgO:PPLN∗
- Control of firing activities in thermosensitive neuron by activating excitatory autapse∗
- Adaptive synchronization of chaotic systems with less measurement and actuation∗
- Dynamics analysis of a 5-dimensional hyperchaotic system with conservative flows under perturbation∗