
QuEChERS前处理HPLC柱后衍生法测定大米粉中克百威含量的研究
2021-12-15 07:42蒋国振
粮食与食品工业 2021年6期
夏 辉,刘 旭,蒋国振,刘 萍,赵 荔
陕西省粮食质量安全中心 (西安 710016)
克百威是一种农业种植中使用的广谱杀虫剂。可用于防治土壤内及地面上的300多种害虫和线虫。克百威对胆碱酯酶抑制的不可逆性决定了其对人、畜、禽等的毒性极高[1]。虽然国家已经禁止使用克百威等氨基甲酸酯类农药,但作为高效广谱杀虫剂,在农业生产中使用仍较广泛,其在粮食中的残留对健康和环境造成严重危害。
目前,报道的关于测定克百威残留量的方法主要有气相色谱法[2]、液相色谱串联质谱法[3-5]。但采用QuEChERS专用盐包前处理技术,HPLC柱后衍生法测定大米粉中克百威含量的方法鲜有报道。用QuEChERS专用盐包前处理技术是近几年发展起来的一种比较新的色谱前处理技术。该前处理方法具有方便、快捷、准确的特点。一般情况下样品前处理只需要几分钟即可完成,大大缩短了前处理试验所需的时间。处理后的样品完全可以满足色谱、质谱等试验的要求。
本研究方法采用QuEChERS专用盐包前处理,HPLC柱后衍生法测定大米粉中克百威的含量,建立的方法方便、准确、灵敏度高、重现性好,为保障粮食质量安全提供了可靠的方法和依据。
1 材料与方法
1.1 主要仪器
高效液相色谱仪,配有自动进样器、FLR检测器和柱后衍生系统,美国Waters公司;AE200型电子天平,瑞士Mettler Toledo公司;超纯水机,美国Milipore公司。
1.2 主要试剂
克百威标准品,浓度100 μg/mL,含量99.8%,中国食品药品检定研究院;甲醇,色谱纯,德国Merck公司;乙腈,色谱纯,德国Merck公司;QuEChERS专用盐包,美国安捷伦公司;SPE净化柱(Florisil-SPE 1 g/6 mL;LC-NH2-SPE 500 mg/6 mL;C181 g/6 mL);磷酸,优级纯;超纯水。
1.3 方法
1.3.1标准工作溶液的配制
移取1.0 mL克百威标准样品于10 mL棕色容量瓶中,用甲醇定容至刻度,摇匀,制备成10 μg/mL的标准工作液;再逐级用甲醇稀释制备成1.0 μg/mL、0.5 μg/mL、0.1 μg/mL、0.05 μg/mL、0.01 μg/mL的标准工作溶液。
1.3.2标准工作曲线
自动进样器吸取2 μL克百威标准工作溶液,注入高效液相色谱仪进行分析,荧光检测器在激发波长330 nm,发射波长465 nm下记录克百威峰面积,以克百威峰面积为纵坐标,以克百威浓度为横坐标,绘制克百威标准曲线。
1.3.3样品前处理
准确称取大米粉样品(粉碎后过40目筛)10 g(精确至0.01 g)于250 mL具塞三角瓶中。加入30 mL蒸馏水,静置30 min,再加入30 mL乙腈,用振荡器200 r/min振荡提取30 min,提取液过滤至装有QuEChERS专用盐包的离心管中,内含1 200 mg无水硫酸镁、400 mg PSA和400 mg C18,涡旋1 min后,4 000 r/min离心5 min后,吸取5 mL上清液于10 mL离心管中,在50 ℃水浴中氮吹蒸发近干,加入2 mL甲醇溶解残余物,涡旋混匀,待净化。
将固相萃取柱用4 mL甲醇-二氯甲烷溶液预淋洗,当液面到达柱筛板顶部时,立即加入上述待净化溶液,用10 mL离心管收集洗脱液,用2 mL甲醇-二氯甲烷溶液涮洗烧杯后过柱,并重复1次,收集洗脱液于50 ℃水浴中氮吹蒸发近干,加入2.5 mL甲醇溶解残余物,涡旋混匀,微孔滤膜过滤,待测。
1.3.4样品测定
自动进样器吸取2 μL样品测试液注入液相色谱仪,荧光检测器在激发波长330 nm,发射波长465 nm下记录克百威峰面积,根据样品测试液的克百威峰面积与标准曲线比较,计算出克百威的浓度。
1.3.5计算结果
试样中克百威含量计算公式如下:
X=C×V×N/m
式中:X为试样中克百威含量,mg/kg;C为从标准曲线得到的待测液中克百威的浓度,μg/mL;V为样品定容体积,mL;N为样品定容后的稀释倍数;m为试样质量,g。
1.3.6仪器条件
色谱柱:C18色谱柱(4.6 mm×150 mm,4 μm);流动相,甲醇:水溶液,梯度洗脱;流速0.2 mL/min;柱温30 ℃;进样量2 μL;荧光检测器,激发波长330 nm,发射波长465 nm。
1.3.7柱后衍生条件
柱后反应单元的温度,80 ℃;水解温度,100 ℃;衍生温度,室温。柱后衍生低压限:0 psi;0.05 mol/L氢氧化钠溶液,流速0.3 mL/min;OPA试剂,流速0.3 mL/min。
2 结果与讨论
2.1 线性结果
以克百威的峰面积为纵坐标,浓度为横坐标进行线性回归分析,得到克百威标准曲线见图1。克百威的线性回归方程(n=5)为:Y=2.83e+005X+4.50e+003(R2=0.999 6)。结果表明:克百威在0.01~1.00 μg/mL范围内具有良好的线性关系。

图1 克百威标准曲线
2.2 检出限和定量限
图2为克百威检出限色谱图。取标准曲线最低点浓度0.01 μg/mL的克百威标准工作液,用甲醇不断稀释,当信噪比为3时,克百威的检出限为0.002 mg/kg;当信噪比为10时,克百威的检出限为0.006 mg/kg。

图2 克百威检出限色谱图
2.3 精密度
图3为克百威色谱图。取浓度为0.1 μg/mL的克百威标准工作液连续进样6次,记录色谱图。色谱峰面积结果见表1。由表1可知,测得克百威峰面积的RSD为 0.12%,结果表明,此方法精密度较好。

表1 精密度试验结果
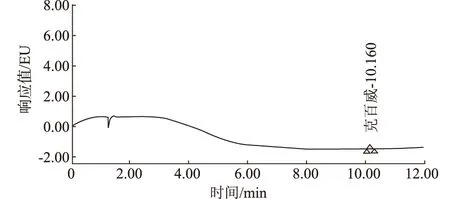
图3 克百威(0.1 μg/mL)精密度试验色谱图
2.4 重复性
取不同加标浓度的样品6份,每份样品称取两个平行样,按1.3.3前处理后,按液相色谱条件做重复性试验,记录色谱图,计算克百威的含量。
由表2可知,样品中克百威的重复性为3.5%~8.3%。结果表明,样品重复性较好。

表2 重复性试验结果
2.5 加标回收率
称取空白样品9份,每份10 g(精确至0.01 g),分别移取浓度为10 μg/mL的克百威标准工作液50 μL、150 μL、500 μL于空白样品中,每组浓度重复3次,配成低、中、高浓度的待测样品加标液,按本试验方法测定,计算加标回收率。克百威加标回收率试验结果见表3,加标试验色谱图见图4。
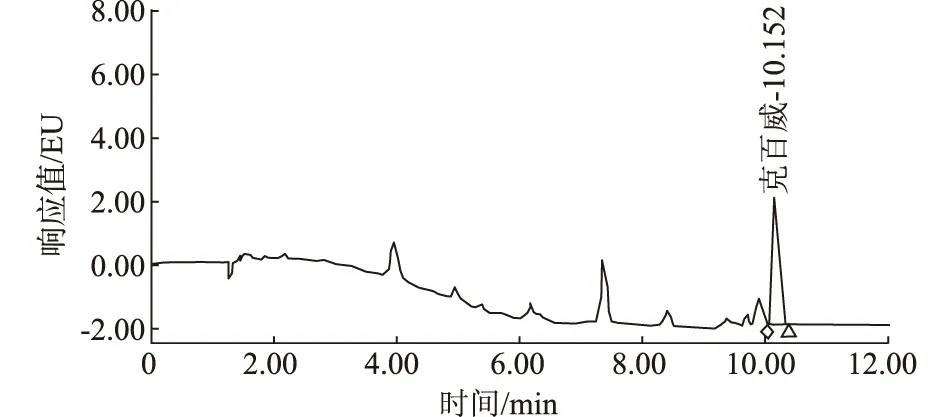
图4 大米粉加标试验色谱图

表3 克百威加标回收率试验结果
由表3可知,克百威低、中、高浓度平均回收率分别为78.7%、84.5%、93.3%,RSD分别为5.3%、4.6%、3.3%,表明该试验方法回收率稳定。
2.6 最佳操作条件的选择
2.6.1提取溶剂的选择
氨基甲酸酯类农药及其代谢物残留量的分析常用甲醇、乙腈、乙酸乙酯、丙酮等有机溶剂[6]。因此,分别选择乙腈:水、甲醇:水、丙酮:水、乙酸乙酯:水作为提取溶剂,溶剂体积比均为1∶1,相同试验条件下,加标浓度为0.50mg/kg。试验结果见表4。

表4 不同溶剂提取效果比较
表4的试验结果表明,使用乙腈∶水(1∶1,v/v)、甲醇∶水(1∶1,v/v)的回收率分别为93.3%和90.2% ,丙酮∶水(1∶1,v/v)、乙酸乙酯∶水(1∶1,v/v)作为提取溶剂的回收率分别为72.5%和65.3% ,前两种选择明显高于后两种。用乙腈∶水(1∶1,v/v)提取的样品基质干扰较小,考虑到综合提取效率、选择性和渗透能力等因素,本方法采用乙腈∶水(1∶1,v/v)作为提取溶剂。
2.6.2固相萃取柱的选择
大米粉主要含有淀粉、蛋白质、脂肪和糖类等物质,基质较复杂。由于克百威极性较大,采用QuEChERS前处理后杂峰较多,须采用固相萃取柱进行净化[7]。主要研究了氨基固相萃取柱、C18固相萃取柱、氟罗里硅土柱的净化情况。在大米粉中添加已知量的克百威标准储备液,采用C18固相萃取柱、氟罗里硅土柱净化的回收率分别为73%和67%,且杂峰较多。氨基固相萃取柱能有效吸附淀粉等糖类物质,回收率较高且干扰较小。
2.6.3流动相的选择
为了获取最优分离效果,考虑到待测溶液最后由甲醇定容,因此根据溶剂的性质,选择研究了甲醇、水不同配比作为流动相的色谱分离情况;研究了梯度洗脱条件对色谱分离效果的影响。结果发现,采用甲醇∶水(1∶9,v/v)作为起始流动相,后逐渐增加有机相比例,当甲醇∶水(7∶3,v/v)时,克百威开始出现色谱峰,在此色谱条件下,克百威的保留时间在10.1~10.2 min,最后逐渐减少甲醇的比例,回到流动相起始比例,可以有效分离克百威。
3 结论
利用QuEChERS专用盐包前处理大米粉等谷物样品,经氨基固相萃取柱纯化,高效液相色谱柱后衍生法测定克百威含量是一种前处理简单、快速、定量准确的检测方法。该方法线性良好,准确度和精密度较高,重复性和回收率均较好,该方法适用于大米粉中克百威含量的测定。
猜你喜欢
理化检验-化学分册(2022年9期)2022-09-22
岭南音乐(2022年4期)2022-09-15
文史春秋(2022年5期)2022-07-18
绿色科技(2021年12期)2021-07-22
科学与财富(2021年34期)2021-05-10
浙江农业学报(2020年11期)2020-12-02
商品与质量(2019年15期)2019-11-28
家教世界·V家长(2019年8期)2019-08-22
